Johnson-Corey-Chaykovsky-Reaktion
Die Johnson-Corey-Chaykovsky-Reaktion, auch Corey-Chaykovsky-Reaktion oder CCR genannt, ist eine chemische Reaktion in der organischen Chemie zur Synthese von Epoxiden, Aziridinen und Cyclopropanen. Sie wurde 1961 von A. William Johnson entdeckt und von E. J. Corey und Michael Chaykovsky weiterentwickelt. Die Reaktion führt unter Zugabe eines Schwefelylids zu einem Keton, Aldehyd, Imin oder Enon zur Bildung des entsprechenden dreigliedrigen Ringsystems. Bei der Reaktion wird diastereoselektiv die trans-Substitution begünstigt, unabhängig von der ursprünglichen Konfiguration. Die Synthese von Epoxiden mit dieser Methode stellt eine Alternative zu der traditionellen Epoxidierung von Olefinen dar.
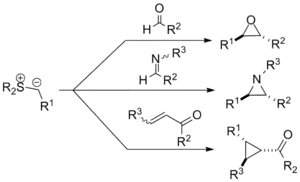
Die Reaktion wird häufig für die Epoxidierung via Methylentransfer eingesetzt und wurde in mehreren bemerkenswerten Totalsynthesen eingesetzt.[1][2][3][4][5]
Geschichte
Die ursprüngliche Veröffentlichung von Johnson beschrieb die Reaktion von 9-Dimethylsulfoniumfluorenylide mit substituierten Benzaldehyd-Derivaten. Die eigentlich angestrebte Wittig-Reaktion fand nicht statt, stattdessen wurde ein Benzalfluorenoxid erhalten. Johnson wies darauf hin, dass die Reaktion zwischen dem Schwefelylid und Benzaldehyd nicht wie die Phosphor. und Arsenyliden zu den Benzalfluorenen führte.[6]
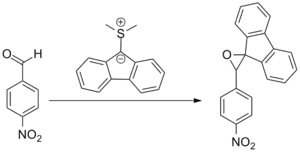
Die anschließende Entwicklung des Dimethyloxosulfoniummethylid (CH3)2SOCH2 und Dimethylsulfoniummethylid (CH3)2SCH2 durch Corey und Chaykovsky, auch bekannt als Corey-Chaykovsky Reagenzien, etablierte die Reaktion als effektive Methylen-Transfer-Reaktion in der organischen Chemie.
Mechanismus
Der Reaktionsmechanismus der Johnson-Corey-Chaykovsky Reaktion besteht im ersten Schritt aus einer nucleophilen Addition des Ylids an eine Carbonyl- oder Imin-Gruppe. Die negative Ladung wird dabei an das Heteroatom übertragen. Das Sulfoniumkation ist eine gute Abgangsgruppe und wird durch Bildung des dreigliedrigen Rings freigesetzt. In der verwandten Wittig-Reaktion, verhindert die Bildung der viel stärkeren Phosphor-Sauerstoff-Doppelbindung die Oxiran-Bildung und stattdessen nimmt erfolgt die Olefinierung über eine viergliedrige cyclische Zwischenstufe.[4]

Die beobachtete trans-Diastereoselektivität resultiert aus der Reversibilität der ersten nucleophilen Addition, die eine Gleichgewichtseinstellung zu Gunsten des bevorzugten anti-Betains gegenüber der Bildung des syn-Betains erlaubt. Durch Addition des Ylid entsteht ein Betain mit benachbarten Ladungen; Berechnungen auf Basis der Dichtefunktionaltheorie haben gezeigt, dass der geschwindigkeitsbestimmende Schritt die Rotation der zentralen Bindung in das Konformere, das die notwendige nucleophile Addition des Sulfonium erlaubt, ist.[1]
_V2.png.webp)
Siehe auch
Einzelnachweise
- Varinder K. Aggarwal, Jeffery Richardson: The complexity of catalysis: origins of enantio- and diastereocontrol in sulfur ylide mediated epoxidation reactions. In: Chemical Communications. Nr. 21, 2003, S. 2644. doi:10.1039/b304625g.
- Varinder K. Aggarwal, Caroline L. Winn: Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis. In: Accounts of Chemical Research. 37, Nr. 8, 2004, S. 611-620. doi:10.1021/ar030045f.
- Yu.G. Gololobov, A.N. Nesmeyanov, V.P. lysenko, I.E. Boldeskul: Twenty-five years of dimethylsulfoxonium ethylide (corey's reagent). In: Tetrahedron. 43, Nr. 12, 1987, S. 2609-2651. doi:10.1016/S0040-4020(01)86869-1.
- An-Hu Li, Li-Xin Dai, Varinder K. Aggarwal: Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement. In: Chemical Reviews. 97, Nr. 6, 1. Oktober 1997, S. 2341-2372. doi:10.1021/cr960411r.
- Eoghan M. McGarrigle, Eddie L. Myers, Ona Illa, Michael A. Shaw, Samantha L. Riches, Varinder K. Aggarwal: Chalcogenides as Organocatalysts. In: Chemical Reviews. 107, Nr. 12, 1. Dezember 2007, S. 5841-5883. doi:10.1021/cr068402y.
- A. William Johnson, Robert B. LaCount: The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides1. In: Journal of the American Chemical Society. 83, Nr. 2, 1. Januar 1961, S. 417-423. doi:10.1021/ja01463a040.