Festphasenmikroextraktion
Festphasenmikroextraktion (englisch solid phase microextraction, SPME) ist eine Methode der Probenahme und Analytenanreicherung, welche sich in einigen Bereichen der chemischen Analytik als vorteilhaft gegenüber klassischen Methoden wie z. B. “Purge and Trap” oder SPE (Solid Phase Extraction, Festphasenextraktion) erwiesen hat. Sie wurde von Janusz Pawliszyn seit 1990 entwickelt.
Weltweit der alleinige Lizenznehmer für Festphasenmikroextraktion-Technologie ist Supelco (U.S. Patent 5,691,206; Europäisches Patent EP523092).
Aufbau eines Probenehmers
Eine schematisierte Abbildung (unten, nicht maßstäblich) gibt den Aufbau eines SPME-Probenehmers wieder.
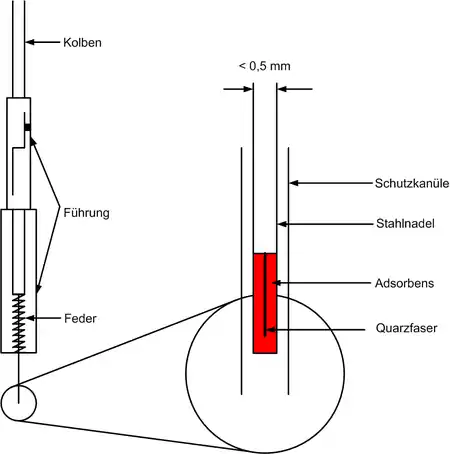
Der Probenehmer besteht im Wesentlichen aus vier Teilen:
- Einer Führung für einen Kolben. Die Führung besitzt einen Bajonett-Verschluss um den Kolben im niedergedrückten Zustand arretieren zu können. Die zwei separaten Teile der Führung können gegeneinander mittels eines Gewindes verstellt werden, um so eine mehr oder weniger große Eindringtiefe der Schutzkanüle in ein Probegefäß zu ermöglichen.
- Einem Kolben zum Ausfahren des Probenehmers. Am Ende des Kolbens befindet sich eine Feder, die den Kolben wieder zurückschiebt, wenn die Arretierung gelöst wird.
- Dem eigentlichen, an den Kolben angeschraubten Probenehmer. Er besteht aus einer Edelstahlnadel, an welcher eine Quarzfaser (“fused silica”) oder Glasfaser („StableFlex“) befestigt ist. Diese Quarzfaser ist mit einer dünnen Schicht Adsorbens überzogen.
- Einer Schutzkanüle, durch die die Faser aufgrund ihrer geringen mechanischen Stabilität geführt wird.
Probenahme
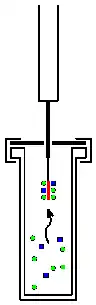
Zuerst wird mit der Schutzkanüle die Durchstichmembran (das Septum) des Probegefäßes durchstochen. Dann wird durch Herabdrücken des Stempels die Faser ausgefahren. Sobald das Adsorbens freiliegt, werden Moleküle des Analyten adsorbiert. Dabei kommt es zu einer starken Anreicherung der Analyten relativ zur Probe. Dabei ist die adsorbierte Menge in erster Näherung linear proportional zur Konzentration in der Probe. In der untenstehenden Abbildung ist die Adsorption aus der Gasphase (“Headspace Sampling”) dargestellt. Analog gelingt dies aber auch mit der Adsorption aus flüssiger Phase (“Immersion Sampling”).
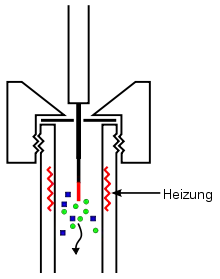
Nach Gleichgewichtseinstellung wird die Arretierung des Kolbens gelöst und die Faser in die Schutzkanüle zurückgezogen, wodurch die Adsorption unterbrochen wird. Dann wird der Probenehmer entfernt. Die adsorbierte Menge verändert sich im eingefahrenen Zustand selbst im Lauf von Stunden kaum, was den einfachen Transport von Proben in ein Labor ermöglicht. Anschließend erfolgt die eigentliche Analyse. Dazu wird der Probenehmer durch das Septum in den Injektor eines Gaschromatographen geschoben. Dann wird die Faser ausgefahren. Aufgrund der hohen Temperatur im Injektor kommt es dann zur Desorption der Analyten. Anschließend wird die Faser wieder eingefahren und der Probenehmer entfernt. Die Desorption kann alternativ durch Extraktion in einem modifizierten Injektorport eines HPLC-Gerätes erfolgen.
Analogie zur Natur
In der Natur arbeitet der Geruchssinn der Schlangen ganz ähnlich: Die Zunge schnellt aus dem Maul, Geruchsstoffe werden von der feuchten Oberfläche adsorbiert, die Zunge wird ins Maul zurückgezogen, die Geruchsstoffe werden an das Jacobson’sche Organ transferiert und erst dort analysiert.
Wiederverwendbarkeit
Bei sorgfältiger Behandlung kann eine Faser ohne Probleme für wenigstens 50 Analysen verwendet werden. Dies setzt voraus, dass die Faser nicht mechanisch (über-)belastet wird, sie regelmäßig durch Ausheizen im Injektor eines Gaschromatographen oder einer geeigneten Konditionierungsstation gereinigt wird und sie nicht mit aggressiven chemischen Lösemitteln, welche das Adsorbens angreifen, in Kontakt kommt.
Probenahmebedingungen
Die Optimierung der Probenahmebedingungen erfordert die Variation verschiedener Parameter. Immer gilt der allgemeine Grundsatz, dass gute Ergebnisse nur durch sorgfältige Einhaltung guter Methoden erreicht werden.
Adsorbens
Kommerziell sind fünf Adsorbentien (in abnehmender Polarität: Polyacrylat, Carbowax, Polydivinylbenzol, Carboxen und PDMS, Polydimethylsiloxan) in mehreren Kombinationen und Schichtdicken für verschiedene Einsatzgebiete verfügbar. Die Faustregel ist, dass polare Analyte mit polaren Adsorbentien und apolare Analyte mit apolaren Adsorbentien analysiert werden sollten. Für hochmolekulare Analyten werden geringere Schichtdicken empfohlen.
Adsorptionsdauer
Die Zeit-Adsorbat-Kurve ist nicht linear, sondern asymptotisch. Für reproduzierbare Messergebnisse sollte nahe der Gleichgewichtseinstellung gearbeitet werden. Wenn verschiedene Analyte in stark wechselnden oder sehr hohen Konzentrationen auftreten, kann es zu Verdrängungserscheinungen auf der Faser kommen. Unter diesen Umständen kann auch im Prä-Äquilibrium gearbeitet werden, man muss dann aber unbedingt auf exakte Zeitkontrolle achten. Bei der Spurenanalytik benötigt man in der Regel längere Adsorptionszeiten.
Adsorptionsbedingungen
Für die Headspace-Analyse von schwerflüchtigen Verbindungen aus festen oder halbfesten Proben wird in der Regel das Probegefäß in einem temperierbaren Heizblock erwärmt. Temperaturangaben in der Literatur variieren von 40–65 °C. Bei der SPME-Analyse von Flüssigkeitsproben kann die Extraktion durch Einstellung eines geeigneten pH-Wertes sowie durch Sättigung mit Kochsalz verbessert werden. Unbedingt erforderlich ist intensives Rühren der Probe (250/min) um die gegenüber der Gasphase niedrigeren Diffusionskoeffizienten auszugleichen. Die Rührgeschwindigkeit muss gleichfalls konstant gehalten werden. Dies gilt sowohl für Messung per Headspace als auch per Immersion.
Desorptionsbedingungen
Die Desorption geschieht in der Regel im Injektor eines Gaschromatographen bei Temperaturen zwischen 200 und 280 °C. Dabei sind fünf Minuten in der Regel ausreichend. Eine Überprüfung kann erfolgen, indem man unter ansonsten konstanten Adsorptionsbedingungen die Desorption verlängert und die Integrale von Analytpeaks gegen die Desorptionsdauer aufträgt. Erhält man eine Parallele zur X-Achse, ist die Desorption vollständig. Für Desorption im Injektorport einer HPLC-Anlage ist analog vorzugehen.
Vorteile
Obwohl man nach dem Obenstehenden anders denken könnte, wird der Zeitaufwand für die Optimierung der Probenahmebedingungen durch die drastische Verkürzung des Zeitaufwandes für die Probenahme mehr als aufgewogen. Außerdem ist eine Automatisierung möglich. Insgesamt wird dadurch für Routineanalysen eine erhebliche Zeitersparnis (> 70 % pro Probe) erreicht.
Der komplette Verzicht auf organische Lösungsmittel bedeutet gegenüber der Flüssig/Flüssig-Extraktion einen erheblichen ökologischen wie ökonomischen Vorteil, da keine anschließende Entsorgung der Lösungsmittel nötig wird. Dazu wird die Gefahr der Querkontamination durch Lösungsmittel beseitigt.
Der Verzicht auf aufwendige Probevorbereitung bedeutet, dass Proben weniger verändert werden. Dies ist besonders bei empfindlichen Analyten von Bedeutung. SPME kann sowohl für gasförmige als auch flüssige Proben verwendet werden. In der Breite gilt dies weder für „Purge and Trap“ oder Flüssig-Flüssig Extraktion. Ein der SPME ähnliches Verfahren stellt die Stir Bar Sorptive Extraction dar.
Nachteile
Aufgrund der geringen Mengen, die von der Faser adsorbiert werden, eignet sich diese Methode, anders als z. B. die SPE, nicht zur präparativen Isolierung der Analyten. Ebenso sind die erzielbaren Anreicherungsfaktoren gegenüber der SPE oder SBSE geringer.
Einsatzgebiete
- Umweltanalytik (z. B. Bestimmung von Herbiziden im Trinkwasser[1] sowie Pestiziden in Blutplasma und Urin[2])
- Lebensmittelanalytik (z. B. Bestimmung von durch Licht erzeugten Abbauprodukten in Milch)
- Aromaanalytik (z. B. von Geosmin,[3] Bestimmung der Geruchsstoffe in Honig und Blütenduft[4] In einem speziellen Fall buchstäblich Erfassung des Duftes einer einzelnen Blüte an Bord des Spaceshuttle)
- Forensik (z. B. Bestimmung von Amphetaminen in Urin[5])
- Probenaufgabe in der Gaschromatographie (spezielle Headspace-Technik)
Literatur
- C. Arthur, J. Pawliszyn: Solid phase microextraction with thermal desorption using fused silica optical fibers. In: Anal. Chem. 62, 1990, S. 2145–2148, doi:10.1021/ac00218a019.
- J. Pawliszyn: Solid Phase Microextraction – Theory and Practice. Wiley-VCH, New York/ Weinheim 1997, ISBN 0-471-19034-9.
Weblinks
- zusätzliche Anwendungsbeispiele (PDF; 2,53 MB)
Einzelnachweise
- Q. Wu, C. Feng, G. Zhao, C. Wang, Z. Wang: Graphene-coated fiber for solid-phase microextraction of triazine herbicides in water samples. In: J Sep Sci. 8. Dez 2011. PMID 22162195
- L. Gao, J. Liu, C. Wang, G. Liu, X. Niu, C. Shu, J. Zhu: Fast determination of paraquat in plasma and urine samples by solid-phase microextraction and gas chromatography-mass spectrometry. In: J Chromatogr B Analyt Technol Biomed Life Sci. 944, 1. Jan 2014, S. 136–140. PMID 24316524
- Z. Ding, S. Peng, Y. Jin, Z. Xuan, X. Chen, L. Yin: Geographical and seasonal patterns of geosmin and 2-methylisoborneol in environmental water in jiangsu province of china. In: Methods Chem. 2014, S. 743924. PMID 25400979
- S. Seisonen, E. Kivima, K. Vene: Characterisation of the aroma profiles of different honeys and corresponding flowers using solid-phase microextraction and gas chromatography-mass spectrometry/olfactometry. In: Food Chem. 169, 15. Feb 2015, S. 34–40. PMID 25236195
- I. Racamonde, R. Rodil, J. B. Quintana, R. Cela: In-sample derivatization-solid-phase microextraction of amphetamines and ecstasy related stimulants from water and urine. In: Anal Chim Acta. 770, 3. Apr 2013, S. 75–84. PMID 23498689