Enzyme-linked Immunosorbent Assay
Enzyme-linked Immunosorbent Assay (ELISA) bezeichnet ein antikörperbasiertes Nachweisverfahren (Assay). Wie der Radioimmunassay (RIA) gehört auch der ELISA zur Gruppe der Immunassay-Verfahren, basiert aber nicht auf einer Radioaktivitätsmessung, sondern auf einer enzymatischen Farbreaktion und gehört somit zu den enzymatischen Immunadsorptionsverfahren (EIA). Das nachzuweisende Antigen wurde ursprünglich über einen Erstantikörper an eine Mikrotiterplatte adsorptiv gebunden und angereichert, ein Enzym-gekoppelter Zweitantikörper (synonym: Detektionsantikörper) führte zur Reaktion eines Farbstoffsubstrates.


Mit Hilfe des ELISA können Proteine (z. B. Antikörper) und Viren, aber auch niedermolekulare Verbindungen wie Hormone, Toxine und Pestizide in einer Probe (Blutserum, Milch, Urin etc.) nachgewiesen werden. Hierbei macht man sich die Eigenschaft spezifischer Antikörper zunutze, die an den nachzuweisenden Stoff (Antigen) binden. Ein Antikörper wird zuvor mit einem Enzym markiert. Die durch das Reporterenzym katalysierte Reaktion dient als Nachweis für das Vorhandensein des Antigens. Das sog. Substrat wird vom Enzym umgesetzt, das Reaktionsprodukt kann üblicherweise durch Farbumschlag, eventuell auch durch Chemolumineszenz nachgewiesen werden. Die Signalstärke ist eine mit einem Photometer sehr genau bestimmbare Funktion der Antigenkonzentration, so dass ELISA als Mehrfachmessungen ausgeführt auch für quantitative Nachweise verwendet werden kann. Als Reporterenzyme werden meistens die Meerrettichperoxidase (HRP, von engl. horseradish peroxidase), die Alkalische Phosphatase (AP) oder seltener auch die Glucose-Oxidase (GOD) verwendet. Im Falle der alkalischen Phosphatase wird als Farbstoffsubstrat (synonym: Chromogen) z. B. p-Nitrophenylphosphat (pNPP) zugegeben, während bei der Peroxidase meistens o-Phenylendiamin (oPD) verwendet wird. Die alkalische Phosphatase spaltet den Phosphatrest vom farblosen Nitrophenylphosphat ab und es entsteht p-Nitrophenol, welches schwach gelb ist. Die Konzentrationsänderung des durch die enzymatische Reaktion entstandenen Farbstoffs kann nach dem Lambert-Beerschen Gesetz mit einem Photometer verfolgt werden. Die Intensität der Farbe steigt dabei mit der Konzentration des entstandenen Nitrophenols und damit auch der Konzentration des zu bestimmenden Antigens in der Probe im Vergleich mit einer Verdünnungsreihe mit bekannten Konzentrationen (Standardreihe).
Geschichte
Der Vorläufer des ELISAs war seit 1960 der Radioimmunoassay.[1] Für einen enzymatischen Nachweis war die direkte Kopplung von Proteinen notwendig, damit das Reportersignal auch nur gekoppelt mit dem spezifisch bindenden Antikörper auftritt. Die chemische Kopplung von Proteinen wurde gleichzeitig von Stratis Avrameas und G. B. Pierce entwickelt.[2] Die Adsorption von Proteinen an Oberflächen war bereits 1966 durch Jerker Porath untersucht worden.[3] Der ELISA wurde im Jahr 1971 gleichzeitig von zwei Arbeitsgruppen entwickelt, darunter Peter Perlmann und Eva Engvall in Schweden.[4][5]
Signalverstärkung
Anstatt eines Enzym-gekoppelten Detektionsantikörpers kann zur Signalverstärkung auch die Kombination eines ungekoppelten Detektions-Antikörpers und eines zusätzlichen (dritten) sekundären Antikörpers (sekundär, weil es ein Antikörper gegen Antikörper ist, engl. secondary antibody), an den ein Enzym gebunden wurde, verwendet werden (s. Abb.). Dies erfordert noch einen weiteren Inkubations- und Waschschritt. Als Puffer wird meistens TBS-T-Puffer verwendet. Obwohl aufwändiger, hat die Verwendung eines sekundären Antikörperkonjugats den Vorteil, dass die kostenaufwändige Herstellung vieler verschiedener Enzym-gekoppelter Primärantikörper, die nur für jeweils ein Antigen spezifisch sind, umgangen werden kann. Die verwendeten sekundären Enzym-gekoppelte Antikörper, die als polyklonale Antikörper gleichzeitig an verschiedene Epitope in der konstanten Region (Fc-Region) von allen Erstantikörpern einer Spezies binden können, sind breiter einsetzbar und führen zu einer Signalverstärkung. Zudem können Sekundärantikörper-Enzym-Konjugate aufgrund der Spezifität für Fc-Regionen eines Antikörpersubtyps von einer Spezies für eine Vielzahl unterschiedlicher Immunassays verwendet werden, sodass es sich bei dem Sekundärantikörper um ein kostengünstigeres industrielles Massenfertigungsprodukt handelt. Eine weitere, häufige Signalverstärkung kann durch die Bindung von Streptavidin- oder Avidin-Konjugaten an biotinylierte Detektionsantikörper im letzten Inkubationsschritt erfolgen. Auch die Detektion mit (Strept-)Avidin-Enzym-Konjugaten führen aufgrund mehrerer Biotinylierungen der Primärantikörper und der daraus folgenden Bindung mehrerer Reportermoleküle zu einer Signalverstärkung.
Moderne Reportersysteme erlauben durch die Verwendung von Fluoreszenz oder der Polymerase-Kettenreaktion teilweise höhere Sensitivitäten (z. B. Immuno-PCR) oder auch parallele Bestimmungen in einem Ansatz (Multiplex), sind aber im strengeren Sinne keine ELISAs.[6]
Antikörper-ELISA
Bei diesem enzymgekoppelten Immunadsorptionstest (EIA) wird das Antigen direkt und ohne einen coating antibody auf die Polystyroloberfläche einer Mikrotiterplatte adsorbiert, wodurch nachfolgend Antikörperkonzentrationen im Vergleich zu einer Standardreihe gemessen werden können.
Sandwich-ELISA

Eine der ELISA-Techniken (Sandwich-ELISA oder auch Antigen-ELISA) verwendet zwei Antikörper (Ak), die beide spezifisch an das nachzuweisende Antigen binden. Hierbei ist es wichtig, dass beide Antikörper an unterschiedlichen Stellen (Epitope) an das Antigen binden, da sie sich sonst gegenseitig behindern würden. Der erste Antikörper (engl. coat antibody oder capture antibody) wird an eine feste Phase (meist Mikrotiterplatten mit 96 wells genannten Vertiefungen) gebunden. Die Probe mit dem nachzuweisenden Antigen wird dann in die wells gegeben und eine Zeit lang inkubiert. Während dieser Zeit bindet der an die Platte gebundene Antikörper das in der Probe vorhandene Antigen. Nach Ablauf der Inkubationsphase wird die Platte gewaschen: Die ungebundenen Bestandteile der Probe werden dadurch entfernt, und zurück bleibt nur das am coat-Antikörper gebundene Antigen. Ein zweiter primärer, unmarkierter Detektionsantikörper wird hinzugefügt, um das Sandwich zu vervollständigen. Durch erneutes Waschen der Platte wird der überschüssige Detektionsantikörper ausgewaschen. Das Ergebnis kann quantifiziert werden, indem ein markierter sekundärer Antikörper hinzugefügt wird, der an den zweiten primären Antikörper bindet und die enzymatische Farbreaktion katalysiert.[7] Für quantitative Nachweise wird üblicherweise eine Serie mit bekannten Antigenkonzentrationen (Standardreihe) durchgeführt, um eine Kalibrierungskurve für das gemessene Signal (optische Extinktion, emittierte Intensität) zu erhalten.
Kompetitiver Immunassay
Häufig wird jedoch auch der kompetitive Immunassay (Enzymgekoppelter Immunadsorptionstest, EIA) angewendet. Hierbei wird zur Detektion kein zweiter, markierter Antikörper verwendet, sondern ein markiertes Kompetitor-Antigen (eine synthetische Verbindung, die dem Analyten strukturell ähnlich ist und auch am Antikörper bindet) eingesetzt. So kommt es zur Kompetition (Konkurrenz) zwischen Analyt und Kompetitor um einen Bindungsplatz am Antikörper. Das Signal verhält sich hier umgekehrt zur Analyt-Konzentration: wenig Analyt = fast alle Antikörperbindestellen werden von markiertem Kompetitor besetzt = starke Farbreaktion; viel Analyt = schwache Farbreaktion. Die verwendeten Nachweissysteme (Enzyme/Substrate) sind meist die gleichen wie beim ELISA.
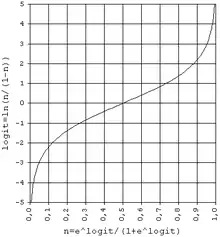
Auswertung des ELISAs mit Hilfe des Logit-Log-Plots
Eine sigmoide Kurvenform tritt dann auf, wenn man auf der x-Achse den Logarithmus der Konzentration und auf der y-Achse die Extinktion (= OD = optische Dichte = Absorption) aufträgt. Diese Darstellungsform ist das halb-logarithmische Diagramm.
Um eine lineare Regression berechnen zu können, muss man diese Sigmoide vorher linearisieren. Zu diesem Zweck behält man die Dimension der x-Achse bei und rechnet die y-Achse in Logit-Werte um. Dabei sollte eine gerade Linie entstehen. Diese Darstellungsform wird Log-Logit-Plot, Logit-Log-Plot oder auch Logit-Plot genannt.
Um Logit-Werte aus den Extinktionswerten berechnen zu können, muss man zuerst die Extinktionswerte (w) normalisieren (n), so dass sie einen Bereich von 0 bis 1 abdecken. Dazu benötigt man die untere (u) und die obere (o) Asymptote der sigmoiden Kurve.

Umkehrfunktion:

Diese normalisierten Extinktionswerte (n) gehen dann in die Logit-Gleichung ein (L):

Umkehrfunktion:

Die Wertepaare des Logit-Log-Plots aus dem x-Wert (= natürlicher Logarithmus der Konzentration) und dem y-Wert (Logit der normalisierten Extinktionswerte (L)) gehen dann in die Berechnung der linearen Regression ein. Diese liefert dann die Höhe (a) und die Steigung (b) der Geradengleichung:

Umkehrfunktion:

Für die Interpolation von unbekannten Messwerten auf die so erstellte Kalibrierkurve, fälschlich auch oft als Eichkurve bezeichnet, benötigt man dann die Umkehrfunktionen. Die höchste Präzision wird in der Nähe des in der Mitte der Sigmoiden liegenden Wendepunktes erreicht, weil an dieser Stelle ihre Steigung am größten ist. Die geringste Genauigkeit entsteht in der Nähe ihrer Asymptoten.
Falls man aus mehreren unterschiedlichen Messwerten mit Hilfe der Asymptoten der Kalibrierungkurve eine Messkurve errechnen kann, dann ist es am genauesten, wenn man die Wendepunktskonzentration der Kalibrierkurve mit der Wendepunktverdünnung der Messkurve vergleicht. Die Asymptoten der Messkurven werden ignoriert, weil man alle Messergebnisse auf den Wendepunkt der Kalibrierkurve beziehen muss, der bei den y-Werten n = 0,5 im halblogarithmischen Diagramm, identisch mit L = 0 im Logit-Log-Plot, zu finden ist. Bei der halblogarithmischen Darstellung der Messkurve dient der natürliche Logarithmus des Kehrwertes der Verdünnung als x-Achse, weil bei den Messwerten die Konzentration vor der Berechnung noch unbekannt ist.
Grundsätzlich ist es auch möglich, anstelle der natürlichen Logarithmen ln die dekadischen Logarithmen oder jene mit der Basis von 2 zu verwenden. Anstelle des Kehrwertes der Verdünnung kann auch nur die Verdünnung (Dilution) selbst eingesetzt werden. Es ist zudem zulässig, den Wert von n als von 0 bis 100 Prozent gehend zu berechnen, sofern man die Gleichungen korrekt modifiziert. Nicht richtig wäre es aber, den Logit direkt aus den nicht normalisierten Extinktionswerten w zu berechnen.
Literatur
- R. A. Goldsby, T. J. Kindt, B. A. Osborne, J. Kuby: Enzyme-Linked Immunosorbent Assay. In: Immunology. 5. Auflage. W. H. Freeman, New York 2003, ISBN 0-7167-4947-5, S. 148–150.
Siehe auch
Einzelnachweise
- R. Yalow, S. Berson: Immunoassay of endogenous plasma insulin in man. In: J. Clin. Invest. 39, Nr. 7, 1960, S. 1157–1175. doi:10.1172/JCI104130. PMID 13846364. PMC 441860 (freier Volltext).
- R. Lequin: Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). In: Clin. Chem. 51, Nr. 12, 2005, S. 2415–2418. doi:10.1373/clinchem.2005.051532. PMID 16179424.
- L. Wide, Jerker Porath: Radioimmunoassay of proteins with the use of Sephadex-coupled antibodies. In: Biochim Biophys Acta (1966) 30: S. 257–260.
- E. Engvall, P. Perlman: Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. In: Immunochemistry. 8, Nr. 9, 1971, S. 871–874. doi:10.1016/0019-2791(71)90454-X. PMID 5135623.
- B. K. Van Weemen, A. H. Schuurs: Immunoassay using antigen-enzyme conjugates. In: FEBS Letters. 15, Nr. 3, 1971, S. 232–236. doi:10.1016/0014-5793(71)80319-8. PMID 11945853.
- S. Leng, J. McElhaney, J. Walston, D. Xie, N. Fedarko, G. Kuchel: Elisa and Multiplex Technologies for Cytokine Measurement in Inflammation and Aging Research. In: J Gerontol a Biol Sci Med Sci. 63, Nr. 8, Oktober 2008, S. 879–884. PMID 18772478. PMC 2562869 (freier Volltext).
- Enzyme-linked Immunosorbent Assay (ELISA) (Ressourcen). Abgerufen am 12. Mai 2018.