Aicardi-Goutières-Syndrom
Aicardi-Goutières Syndrom (AGS) ist eine autosomal-rezessive Erbkrankheit, die erstmals 1984 von den französischen Ärzten Jean François Aicardi und Françoise Goutières beschrieben wurde. Abgegrenzt werden muss das Aicardi-Syndrom, das eine gänzlich andere erbliche Hirnentwicklungsstörung darstellt.
| Klassifikation nach ICD-10 | |
|---|---|
| G93.4 | Enzephalopathie nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Das Aicardi-Goutières-Syndrom ist eine genetisch heterogene Hirnveränderung (Enzephalopathie), die klinisch Ähnlichkeiten mit einer intrauterin erworbenen Infektion aufweist, jedoch ohne Erregernachweis. Vielmehr liegt eine genetische Ursache zugrunde, bei der Zellkern-Enzyme vermindert aktiv sind, die die Chromosomen von fälschlich eingebauten RNA-Proteinen „säubern“. Durch die verminderte Enzymaktivität reichern sich DNA-Abschnitte in der Zelle an, die dadurch zugrunde geht und eine von der Immunabwehr vermittelte Entzündung auslöst.
Das Syndrom kann auch zu den Leukodystrophien eingeordnet werden, die mit einer Störung der Myelinisierung verbunden sind.
Bisher sind kaum hundert Fälle beschrieben worden.
Symptome
Die betroffenen Kinder fallen zumeist durch Schwierigkeiten beim Füttern, ruckartige Augenbewegungen, gelegentliche leichte Fieberschüben, Erbrechen und Zappeligkeit auf. Bei ca. einem Drittel der Patienten kommt es im Alter von sechs Monaten zum Verlust vorher gelernter motorischer Fähigkeiten. Die Kinder zeigen spastische Lähmungen oder dystone, unkoordinierte Bewegungen. Die Spastizität und Bewegungsstörungen führen oft zu Kontrakturen an Armen und Beinen. Gelegentlich treten Krampfanfälle auf. Es kommt zu einer zunehmenden psychomotorischen Retardierung. Viele Patienten versterben in der frühen Kindheit.
In einer Untersuchung[1] an elf italienischen Kindern traten die ersten Symptome im Mittel nach 3,3 Monaten auf, meist mehrere Symptome gleichzeitig: Je fünfmal Irritabiliät und psychomotorische Entwicklungsstörung, je viermal Fieberschübe und Schluckstörungen sowie viermal Muskeltonusstörungen (Hypo- und Hypertonie), bei einem Kind Anfälle und eine Vergrößerung von Leber und Milz.
In der Untersuchung des Hirnwassers (Liquor cerebrospinalis) zeigt sich eine Erhöhung der weißen Blutkörperchen (CSF-Lymphozytose) und des Alpha-Interferons als Hinweis auf eine entzündliche Ursache. Im Blut finden sich eine Verminderung der Blutplättchen (Thrombozytopenie) und ein Anstieg der Leber-Enzymwerte (Leber-Transaminasen). Oft sind Leber und Milz vergrößert.
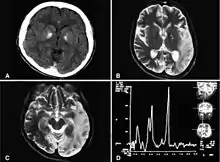
In einer Schnittbilduntersuchung des Kopfes (Computertomographie, Magnetresonanztomographie) ist ein Hirnsubstanzverlust (Atrophie) nachweisbar, sowie eine Fehlbildung der weißen Hirnsubstanz (Leukodystrophie). Hinzu kommen zahlreiche Verkalkungsherde. Daher wird die Erkrankung auch als Basalganglien-Enzephalopathie oder kalzifizierende Enzephalopathie mit intrakranialer Verkalkung und chronischer CSF-Lymphozytose bezeichnet.
Da gelegentlich Hautveränderungen, ein Komplementfaktormangel und antinukleäre Antikörper nachgewiesen wurden, wurde ein Zusammenhang mit rheumatischen Erkrankungen vermutet.
Wegen der Ähnlichkeit mit einer intrauterinen Infektion mit Toxoplasma-Parasiten (Toxoplasmose) wurde das Syndrom synonym auch als Pseudotoxoplasmose-Syndrom bezeichnet.
Lokalisation der Genmutation
Inzwischen wurden fünf Genorte lokalisiert (TREX1, RNASEH2A, RNASEH2B, RNASEH2C, und SAMHD1), die dieses Syndrom verursachen können.[2] Zuerst wurde in fünf nicht verwandten Familien eine Mutation des Gens TREX1 auf Chromosom 3p21 beschrieben.[3][4]
Bei den Genen RNASEH2-A, -B und -C handelt es sich um die Genloci für die drei Proteine, die zusammen das trimere Zellkern-Enzym Ribonuklease H2 (RNase H) bilden. Dieses ist für die Entfernung fälschlicherweise in die DNA eingebauter RNA-Moleküle zuständig, was physiologisch regelmäßig vorkommt. Die RNA-Moleküle sind wesentlich anfälliger für Schädigungen als die normalen DNA-Moleküle mit erhöhter Geninstabilität bis hin zur Hydrolyse mit Störung der Erbinformation. Ist die RNase H komplett ausgeschaltet, führt dies bei der Knockout-Maus zur frühembryonalen Letalität durch Unterbrechung des Zellzyklus bereits während der Gastrulation. Bei teilweise verminderter Enzymaktivität reichern sich Gen-Bruchstücke in den Zellen an und lösen eine p53-vermittelte Unterbrechung des Zellzyklus oder eine Apoptose ein, was dann zu einer Entzündungsreaktion durch die angeborene Immunantwort führt, die einer Autoimmunreaktion wie beim Systemischen Lupus erythematodes gleicht.[5]
Einzelnachweise
- G. Lanzi u. a.: Neurology. Band 64, 2005, S. 1621–1624 (Erste Symptome bei elf italienischen Kindern).
- Aicardi-Goutières-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- Y. J. Crow u. a.: Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. In: Nature Genet. Band 38, 2006, S. 917–920, PMID 16845398 (Erstbeschreibung der TREX1-Mutation).
- G. Ramantani, J. Kohlhase u. a.: Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. In: Arthritis and Rheumatism. Band 62, Nummer 5, Mai 2010, S. 1469–1477,doi:10.1002/art.27367.
- Martin A. M. Reijns u. a.: Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. In: Cell. Band 149, 25. Mai 2012, S. 1008–1022, doi:10.1016/j.cell.2012.04.011.