Zellseneszenz
Zelluläre Seneszenz (von lateinisch senescere = „alt werden, altern“) ist ein Phänomen, bei dem Zellen aufhören, sich zu teilen. In ihren Experimenten aus den frühen 1960er Jahren fanden Leonard Hayflick und Paul Moorhead heraus, dass normale menschliche fetale Fibroblasten in Kultur maximal etwa 50 Zellpopulationsverdopplungen erreichen, bevor sie altern[1][2][3]. Dieses Phänomen wird als replikative Seneszenz oder Hayflick-Grenze bezeichnet. Hayflicks Entdeckung, dass normale Zellen sterblich sind, stürzte ein 60-jähriges Dogma in der Zellbiologie, das besagt, dass alle gezüchteten Zellen unsterblich sind. Hayflick fand heraus, dass die einzigen unsterblichen Kulturzellen Krebszellen sind[4]. Die zelluläre Seneszenz wird häufig durch Tumorsuppressor-Mechanismen angestoßen, die inaktiviert werden müssen, um ein Fortschreiten in Richtung Seneszenz zu verhindern und dadurch eine Krebsentstehung zu ermöglichen. Durch die Überwindung oder Umgehung dieser Tumorsuppressor-Mechanismen entziehen sich Krebszellen der Kontrolle des Zellzyklus, was zu genomischer Instabilität und unkontrollierter Proliferation führt. MicroRNAs (miRNAs) haben sich als wesentliche Faktoren herausgestellt, die zur zellulären Seneszenz beitragen oder diese verhindern.[5]
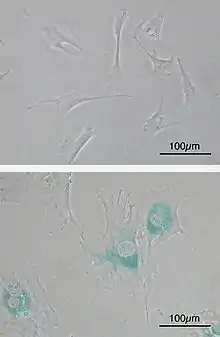
(Unten) MEFs, gealtert nach weiteren Passagen in Zellkultur. Die Zellen sind größer, flacher und exprimieren die Seneszenz-assoziierte β-Galaktosidase (SABG, blaue Bereiche), einen Marker für zelluläre Seneszenz.
Zelluläre Mechanismen
Mechanistisch kann zelluläre Seneszenz durch eine Veränderung der DNA ausgelöst werden, die sich aus der Verkürzung der Telomere bei jedem Zellteilungsvorgang ergibt. In diesem Spezialfall spricht man von replikativer Seneszenz. Zellen können jedoch auch unabhängig von der Anzahl der Zellteilungen zur Seneszenz gebracht werden, zum Beispiel über DNA-Schäden als Reaktion auf erhöhte reaktive Sauerstoffspezies (ROS), durch Aktivierung von Onkogenen und durch Zell-Zell-Fusion. Die Anzahl der alternden Zellen im Gewebe steigt während der normalen Alterung deutlich an.[6] Obwohl sich alternde Zellen nicht mehr vermehren können, bleiben sie metabolisch aktiv und nehmen im Allgemeinen einen immunogenen Phänotyp an, der u. a. aus einem pro-inflammatorischen Sekretom und der Hochregulation von Immunliganden besteht. Seneszenz-assoziierte β-Galaktosidase gilt zusammen mit CDK-Inhibitor 2A (CDKN2A) (cyclin dependent kinase inhibitor 2A, auch bekannt als p16) als guter Biomarker für zelluläre Seneszenz. Dies führt dennoch zu falsch positiven Signalen, da auch reifende Gewebe-Makrophagen seneszenz-assoziierte β-Galaktosidase zeigen, genauso wie T-Zellen CDKN2A exprimieren.[6] Die DNA-Schadensantwort (DNA Damage Response, DDR) blockiert den Verlauf des Zellzyklus, bis Schäden, wie beispielsweise Doppelstrangbrüche (DSBs), repariert sind. Seneszente Zellen zeigen persistente Stellen mit DNA-Schäden, die resistent gegen endogene DNA-Reparaturaktivitäten zu sein scheinen. Seneszente Zellen in Kultur und im Gewebe von gealterten Säugetieren bewahren ihre DSBs, die mit DDR-Markern assoziiert sind. Es wird vermutet, dass diese DSBs wichtige Treiber des Alternsprozesses sind.[7]
Rolle der Telomere
In letzter Zeit hat die Rolle der Telomere bei der zellulären Seneszenz Interesse geweckt, insbesondere im Hinblick auf mögliche Nebenwirkungen des Klonens. Mit jedem Zellzyklus verkürzen sich die chromosomalen Telomere sukzessiv, wodurch die Anzahl der Teilungen der Zelle begrenzt werden kann. Dieser Vorgang trägt zur Alterung bei. Die Telomerverkürzung verändert zum Beispiel auch die Mechanismen beim alternativen RNA-Spleißen. Dadurch werden Toxine wie Progerin produziert, die einen Alterungsprozess durch Abbau und Funktionseinschränkungen von Geweben induzieren.[8] So wird (etwa beim Hutchinson-Gilford-Progerie-Syndrom) das bei fehlerhaftem Spleißen statt dem Lamin A entstandene Progerin dauerhaft an die Kernmembran einer Zelle gebunden, und es kommt zu veränderter Kernmorphologie, erhöhtem DNA-Schaden, epigenetischen und metabolischen Veränderungen, transkriptioneller Dysregulation, Verlust der Proteinhomöostase, Funktionsstörung der Stammzellen, beschleunigter Seneszenz und Zelltod.[9]
Weitere Merkmale seneszenter Zellen
Ein Senescence Associated Secretory Phenotype (SASP) bestehend aus entzündlichen Zytokinen, Wachstumsfaktoren und Proteasen ist ein weiteres charakteristisches Merkmal seneszenter Zellen.[10] Der SASP ist mit vielen altersbedingten Krankheiten verbunden, darunter Typ-2-Diabetes und Atherosklerose[6]. Dies hat Forscher motiviert, senolytische Medikamente (Senolytika) zu entwickeln, um seneszente Zellen abzutöten bzw. zu eliminieren, um die Gesundheit älterer Menschen zu verbessern[6]. Deren positive Wirkung wurde bereits bei Mäusen gezeigt.[11] Der Kern der alternden Zellen ist gekennzeichnet durch seneszenzassoziierte Heterochromatin-Foci (SAHF) und DNA-Segmente mit Chromatinveränderungen, die die Seneszenz verstärken (DNA-SCARS).[12] Seneszente Zellen ermöglichen die Tumorunterdrückung, die Wundheilung und die embryonale / plazentare Entwicklung, obwohl sie eine pathologische Rolle bei altersbedingten Erkrankungen spielen.[13]
Organismen ohne Seneszenz
Zelluläre Seneszenz wird nicht bei allen Organismen beobachtet. In den Arten, in denen zelluläre Seneszenz beobachtet wird, werden die Zellen post-mitotisch, das heißt, sie replizieren nicht mehr durch den Prozess der Mitose. Sie sind damit in einem Zustand replikativer Seneszenz. Wie und warum einige Zellen bei einigen Arten post-mitotisch werden, war Gegenstand vieler Forschungen und Spekulationen, es wird aber angenommen, dass sich zelluläre Seneszenz u. a. entwickelt hat, um den Beginn und die Ausbreitung von Krebs zu verhindern. Somatische Zellen, die sich häufig geteilt haben, weisen mehr DNA-Mutationen auf und laufen daher Gefahr, sich zu Krebszellen zu entwickeln, wenn sich die Teilung fortsetzt. Die alternden Zellen scheinen zudem einen immunogenen Phänotyp (s.o) zu entwickeln, der es dem Immunsystem ermöglicht, sie zu erkennen und zu eliminieren.[14]
Siehe auch
Einzelnachweise
- M. Collado, M. A. Blasco, M. Serrano: Cellular senescence in cancer and aging. In: Cell. Band 130, Nummer 2, Juli 2007, S. 223–233, doi:10.1016/j.cell.2007.07.003, PMID 17662938 (Review).
- M Hayat: Tumor dormancy, quiescence, and senescence, Volume 2: Aging, cancer, and noncancer pathologies. Springer, 2014, S. 188.
- T Tollefsbol: Epigenetics of Aging. Springer, 2010, ISBN 978-1-4419-0638-0, S. 227.
- J. W. Shay, W. E. Wright: Hayflick, his limit, and cellular ageing. In: Nature reviews. Molecular cell biology. Band 1, Nummer 1, 10 2000, S. 72–76, doi:10.1038/35036093, PMID 11413492.
- M. Neault, F. Couteau u. a.: Molecular Regulation of Cellular Senescence by MicroRNAs: Implications in Cancer and Age-Related Diseases. In: International review of cell and molecular biology. Band 334, 2017, S. 27–98, doi:10.1016/bs.ircmb.2017.04.001, PMID 28838541 (Review).
- B. G. Childs, M. Durik u. a.: Cellular senescence in aging and age-related disease: from mechanisms to therapy. In: Nature medicine. Band 21, Nummer 12, Dezember 2015, S. 1424–1435, doi:10.1038/nm.4000, PMID 26646499, PMC 4748967 (freier Volltext) (Review).
- Asao Noda,corresponding author Shuji Mishima, Yuko Hirai, Kanya Hamasaki, Reid D. Landes, Hiroshi Mitani, Kei Haga, Tohru Kiyono, Nori Nakamura, and Yoshiaki Kodama: Progerin, the protein responsible for the Hutchinson-Gilford progeria syndrome, increases the unrepaired DNA damages following exposure to ionizing radiation. In: PMC. 1. Oktober 2015, abgerufen am 15. September 2021 (englisch).
- K. Cao, C. D. Blair, D. A. Faddah, J. E. Kieckhaefer, M. Olive, M. R. Erdos, E. G. Nabel, F. S. Collins: Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. In: The Journal of clinical investigation. Band 121, Nummer 7, Juli 2011, S. 2833–2844, doi:10.1172/JCI43578, PMID 21670498, PMC 3223819 (freier Volltext).
- Davor Lessel, Christian Kubisch: Genetisch bedingte Syndrome mit Zeichen einer vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 116, Heft 29 f., 22. Juli 2019, S. 489–496, hier: S. 491–493.
- N. Malaquin, A. Martinez, F. Rodier: Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. In: Experimental Gerontology. Band 82, 09 2016, S. 39–49, doi:10.1016/j.exger.2016.05.010, PMID 27235851 (Review).
- A. R. Mendelsohn, J. W. Larrick: Cellular Senescence as the Key Intermediate in Tau-Mediated Neurodegeneration. In: Rejuvenation research. Band 21, Nummer 6, Dezember 2018, S. 572–579, doi:10.1089/rej.2018.2155, PMID 30489222.
- F. Rodier, J. Campisi: Four faces of cellular senescence. In: Journal of Cell Biology. Band 192, Nummer 4, Februar 2011, S. 547–556, doi:10.1083/jcb.201009094, PMID 21321098, PMC 3044123 (freier Volltext) (Review).
- D. G. Burton, V. Krizhanovsky: Physiological and pathological consequences of cellular senescence. In: Cellular and molecular life sciences : CMLS. Band 71, Nummer 22, November 2014, S. 4373–4386, doi:10.1007/s00018-014-1691-3, PMID 25080110, PMC 4207941 (freier Volltext) (Review).
- D. G. Burton, R. G. Faragher: Cellular senescence: from growth arrest to immunogenic conversion. In: Age. Band 37, Nummer 2, 2015, S. 27, doi:10.1007/s11357-015-9764-2, PMID 25787341, PMC 4365077 (freier Volltext) (Review).