Makrodaktylie
Unter Makrodaktylie (griech. μακρός makros „groß“, „lang“, δαϰτυλος dáktylos „Finger“; „Zehe“) versteht man ein gegenüber den anderen überproportionales Wachstum, einen Riesenwuchs (Gigantismus, Hypersomie) eines oder mehrerer Finger oder Zehen.
| Klassifikation nach ICD-10 | |
|---|---|
| Q74.0 | Sonstige angeborene Fehlbildungen der oberen Extremität(en) und des Schultergürtels |
| Q74.2 | Sonstige angeborene Fehlbildungen der unteren Extremität(en) und des Beckengürtels |
| ICD-10 online (WHO-Version 2019) | |
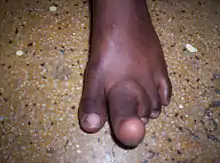
Zur Abgrenzung gegenüber anderen Erkrankungen wie z. B. Gefäßmissbildung, Lymphangiom, Tumoren, Exostosen, bei denen einzelne Komponenten der Finger- bzw. Zehenanatomie betroffen sind, sind bei der Makrodaktylie alle Gewebe in proportionaler Weise vergrößert.[1]
Das Proteus-Syndrom kann als Sonderform betrachtet werden.[1]
Erworbene Makrodaktylie
Erworbene Formen sind selten.[2]
Mögliche Ursachen sind:
- Entzündung, z. B. bei Infektion
- Tumoren wie Osteoid-Osteom, Melorheostose, Lipofibromatöses Hamartom[3][4]
- Arteriovenöse Malformation vor Schluss der Epiphysenfuge
- Elephantiasis
- Still-Syndrom
- Amyloidose
- Akromegalie
- Tuberöse Sklerose[5]
Isolierte Makrodaktylie
Diese angeborene Form als Kongenitale Makrodaktylie ist die häufigste. An der Hand sind ein oder mehrere Finger typischerweise innerhalb des Versorgungsgebietes eines überlangen Nervens vergrößert und verdickt. Der Zeigefinger ist am häufigsten betroffen. Bei mehreren Fingern sind diese benachbart.[6][7]
Als Ursache wird, wie bei den meisten angeborenen Extremitätenfehlbildungen, Störung während der 4. bis 6. Schwangerschaftswoche angenommen.[8]
Bislang wurden Mutationen im PIK3CA-, AKT1- und PTEN-Gen als Auslöser gefunden.[9]
Im Rahmen von Erkrankungen
Bei folgenden Erkrankungen und Syndromen kann es zu Makrodaktylien kommen:[10]
- Hochwuchs-Skoliose-Makrodaktylie der Großzehen-Syndrom, Synonym: Epiphyseal chondrodysplasia, Miura type[11]
- Fettriesenwuchs (progressive Makrodystrophia lipomatosa)
- Hemihypertrophie
- Klippel-Trénaunay-Weber-Syndrom
- Lhermitte-Duclos-Syndrom
- Lipodystrophie Typ Berardinelli
- Lipofibromatose
- Maffucci-Syndrom
- Neurofibromatose
- Ruvalcaba-Myhre-Smith-Syndrom
Klinische Erscheinungen
Die Vergrößerung kann bereits bei Geburt vorliegen und sich proportional zum Wachstum entwickeln oder erst nach den ersten Lebensjahren auftreten und dann ein rascheres Wachstum aufweisen[12].
Bevorzugt sind die mittleren Strahlen betroffen. Häufiger findet man eine Syndaktylie. Am Fuß steht das Problem der Schuhversorgung im Vordergrund, auch die verdrängende Wirkung auf benachbarte Zehen.
Diagnostik
Zur Abgrenzung der aufgeführten Differentialdiagnosen ist eine Röntgenaufnahme erforderlich. Die knöchernen Strukturen sind außer ihrer Übergröße nicht verändert.
Behandlung
Die Behandlung richtet sich nach dem Ausmaß der Beeinträchtigung und reicht von der Epiphyseodese bis zur teilweisen Amputation. Sie erfolgt in Anbindung an eine kinderorthopädische Fachabteilung.[13]
Literatur
- J. Wu, G. Tian, Y. Ji, J. P. Higgins, W. P. Lee: Clinical Characteristics of 90 Macrodactyly Cases. In: The Journal of hand surgery. Band 45, Nummer 10, Oktober 2020, S. 982.e1–982.e5, doi:10.1016/j.jhsa.2020.03.002, PMID 32299688.
- A. Gupta, C. S. Burke: Macrodactyly. In: J. Abzug, S. Kozin und D. Zlotolow (Hrsg.): The Pediatric Upper Extremity. 2015, https://doi.org/10.1007/978-1-4614-8515-5_16
- R. R. van Rijn, J. G. Blickman: Differenzialdiagnosen in der pädiatrischen Radiologie. Thieme 2012, ISBN 978-3-13-169571-0.
- Lexikon Orthopädie – Makrodaktylie
Einzelnachweise
- R. Hoffmann, H. Krimmer: Makrodaktylie. In: R. Hoffmann (Hrsg.): Checkliste Handchirurgie, 4. Auflage, 2016. doi:10.1055/b-004-138006, eref.Thieme
- I. G. Vetrano, L. M. Sconfienza, D. Albano, V. Chianca, V. Nazzi: Recurrence of carpal tunnel syndrome in isolated non-syndromic macrodactyly: DTI examination of a giant median nerve. In: Skeletal radiology. Band 48, Nummer 6, Juni 2019, S. 989–993, doi:10.1007/s00256-018-3098-y, PMID 30343441.
- A. Razzaghi, D. J. Anastakis: Lipofibromatous hamartoma: review of early diagnosis and treatment. In: Canadian journal of surgery. Journal canadien de chirurgie. Band 48, Nummer 5, Oktober 2005, S. 394–399, PMID 16248139, PMC 3211895 (freier Volltext) (Review).
- M. Amouyel-Castier, V. Goëb, H. Deramond, B. Bonnaire: A relapsing macrodactyly. In: Clinical Rheumatology. Band 34, Nummer 4, April 2015, S. 803–805, doi:10.1007/s10067-014-2580-8, PMID 24687379.
- B. Sahoo, S. Handa, B. Kumar: Tuberous sclerosis with macrodactyly. In: Pediatric dermatology. Band 17, Nummer 6, 2000 Nov-Dec, S. 463–465, doi:10.1046/j.1525-1470.2000.01821.x, PMID 11123780.
- Makrodaktylie der Finger. In: Orphanet (Datenbank für seltene Krankheiten).
- Makrodaktylie der Zehen. In: Orphanet (Datenbank für seltene Krankheiten).
- Carl Joachim Wirth, Abdul Kader Martini: Orthopädie und orthopädische Chirurgie. Georg Thieme Verlag, 2003, ISBN 978-3-13-126211-0, S. 216–.
- W. Tian, Y. Huang, L. Sun, Y. Guo, S. Zhao, M. Lin, X. Dong, W. Zhong, Y. Yin, Z. Chen, N. Zhang, Y. Zhang, L. Wang, J. Lin, Z. Yan, X. Yang, J. Zhao, G. Qiu, J. Zhang, Z. Wu, N. Wu: Phenotypic and genetic spectrum of isolated macrodactyly: somatic mosaicism of PIK3CA and AKT1 oncogenic variants. In: Orphanet Journal of Rare Diseases. Band 15, Nummer 1, 10 2020, S. 288, doi:10.1186/s13023-020-01572-9, PMID 33054853, PMC 7556951 (freier Volltext).
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Hochwuchs-Skoliose-Makrodaktylie der Großzehen-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- F. Hefti: Kinderorthopädie in der Praxis. Springer 1998, ISBN 3-540-61480-X, Seite 492
- C. J. Jacobs, M. Vreeburg, C. E. de Die-Smulders, H. M. Staal, T. Lauwers: Surgical management for isolated macrodactyly in an adult PIK3CA mutant. In: JPRAS open. Band 26, Dezember 2020, S. 86–90, doi:10.1016/j.jpra.2020.10.002, PMID 33251316, PMC 7680883 (freier Volltext).