Lennox-Gastaut-Syndrom
Das Lennox-Gastaut-Syndrom (LGS), auch unter dem Synonym Lennox-Syndrom bekannt, ist eine meist schwer behandelbare Form von Epilepsie, die bei Kindern in der Regel in der Zeit zwischen dem zweiten und sechsten Lebensjahr beginnt, mit häufigen und verschiedenen Anfallstypen einhergeht und deren Ursache in einer ursächlich vielfältigen Schädigung des Gehirns besteht, die entweder vorgeburtlich (pränatal), während der Geburt (perinatal) oder nachgeburtlich (postnatal) entstanden ist.
| Klassifikation nach ICD-10 | |
|---|---|
| G40.4 | Sonstige generalisierte Epilepsie und epileptische Syndrome Lennox-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Geschichte
Benannt wurde das Syndrom nach dem US-amerikanischen Neurologen und Epileptologen William G. Lennox (Boston/USA) und dem französischen Neuroanatom, Neurologen, klinischen Neurophysiologen und Epileptologen Henri Gastaut (Marseille/Frankreich), die diese Form der Epilepsie in den 1950er und 1960er Jahren erstmals eingehend unter wissenschaftlichen Gesichtspunkten beschrieben und sich intensiv mit ihrer Erforschung und Abgrenzung zu anderen Formen der Epilepsie befassten.[1][2][3] Gastaut stützte sich dabei u. a. auch auf die Doktorarbeit seiner Mitarbeiterin Charlotte Dravet.[4]
Auftretenshäufigkeit
Schätzungsweise 5 von 100 Kindern mit Epilepsie haben ein LGS, wobei Jungen prozentual gesehen häufiger betroffen sind als Mädchen. Während ein Teil der Kinder vor dem Auftreten der Anfälle keine Auffälligkeiten zeigte, hatte der andere Teil bereits eine Form von Epilepsie, die dann in das LGS übergegangen ist. Bei durchschnittlich einem von fünf Kindern mit LGS war vor dessen Auftreten ein West-Syndrom (symptomatisch) diagnostiziert worden, das durch BNS-Krämpfe kennzeichnet und nicht selten innerhalb des zweiten Lebensjahres in ein LGS übergeht.
Ursache
Die Ursache dieser Form von Epilepsie muss im Einzelfall betrachtet werden, da es keine einheitliche Ursache gibt: Bei einem von fünf Kindern entsteht das LGS aus dem West-Syndrom. Auch sonst sind in der Anamnese auffallend häufig Angaben über Neugeborenenkrämpfe oder fokale und generalisierte Anfälle zu finden.
Bei bis zu zwei Dritteln der Kinder mit LGS tritt die Epilepsie als Folge bzw. Symptom einer Gehirnschädigung (Enzephalopathie) oder einer anderen Erkrankung bzw. Entwicklungsstörung auf. Häufige Ursachen sind beispielsweise Tuberöse Sklerose (Bourneville-Syndrom), Stoffwechselkrankheiten, Gehirnentzündungen wie z. B. Enzephalitis, Meningitis oder Toxoplasmose, tief greifende hirnorganische Störungen wie sie z. B. durch Sauerstoffmangel unter der Geburt oder aufgrund einer Frühgeburt entstehen können sowie Schädel-Hirn-Traumata verschiedener Art. Lässt sich eine solche oder eine ähnliche Ursache nachweisen, spricht man von einem symptomatischen Lennox-Gastaut-Syndrom, da die Anfälle als Begleiterscheinung oder Merkmal (Symptom) einer anderen Besonderheit auftreten.
Bei bis zu einem Drittel der Fälle kann keine Grunderkrankung nachgewiesen werden, die augenscheinlich zum Auftreten des LGS geführt hat. In solchen Fällen spricht man von einem kryptogenen bzw. idiopathischen Lennox-Gastaut-Syndrom.
Merkmale
In der Regel treten epileptische Anfälle im Rahmen des LGS zwischen dem zweiten und sechsten Lebensjahr erstmals auf, wobei in Ausnahmefällen der Zeitpunkt der Erstmanifestation auch innerhalb des zweiten Lebensjahres oder nach dem achten Lebensjahr liegen kann. Das Erscheinungsbild weist deutliche Parallelen zum West-Syndrom auf, sodass eine Verwandtschaft wahrscheinlich ist.
Typisch für das LGS ist das täglich mehrmalige Auftreten von Anfällen. Auffallend ist auch die große Spannbreite der vorkommenden Anfallstypen, die in dieser Vielfalt bei keinem anderen Epilepsie-Syndrom vorkommt: Am häufigsten sind tonische Anfälle in unterschiedlicher Ausprägung, die oft im Schlaf auftreten (bei etwa neun von zehn Kindern), am zweithäufigsten sind myoklonische Anfälle zu beobachten, die gehäuft bei Müdigkeit auftreten.
Daneben können atonische Anfälle, atypische Absencen, fokale und teilweise auch generalisierte tonisch-klonische (Grand-mal-) Anfälle auftreten. Darüber hinaus kommt es bei einem von zwei Kindern zu einem epileptischen Status (Status epilepticus), meist in Form eines nichtkonvulsiven Status, der sich durch Verwirrtheit, Apathie und fehlende Reaktionen kennzeichnet. Insbesondere die atypischen Absencen können mit statusartiger Häufung vorkommen. Vielfach kommt es durch die Anfälle zu plötzlichen Stürzen (bei tonischen, atonischen und myoklonischen Krämpfen) bzw. auffallenden Haltungsverlusten, weshalb manche betroffene Kinder einen Sturzhelm zum Schutz des Kopfes tragen.
Kinder mit dem LGS zeigen häufig eine deutliche Verzögerung der körperlichen Gesamtentwicklung, eine kognitive Behinderung und Verhaltensauffälligkeiten.
Diagnose
Das LGS lässt sich als Krankheitsbild oft nur schwer eingrenzen bzw. eindeutig von anderen Krankheitsbildern abgrenzen, da es diverse und zum Teil fließende Übergänge zu ähnlichen Syndromen gibt. Wie schon erwähnt gibt es keine einheitliche Ursache, die die Diagnostik vereinfachen würde.
Offen sichtbar ist diese Form der Epilepsie durch häufige und vielfältige Anfälle. Bei der Messung der Gehirnströme mittels eines EEGs zeigt sich ein in der Regel verlangsamter Grundrhythmus mit stets langsamem Spike-wave-Muster oder multifokalen und generalisierenden Sharp-slow-wave-Entladungen, die mit einer Häufigkeit zwischen 1,5 und 2,5 mal pro Sekunde registriert werden können. Veränderungen der Muster sind dahingehend möglich, dass es zu Seitenunterschieden und herdförmigen Besonderheiten in der Ableitung kommen kann. Im Schlaf können häufig tonische Muster, also Serien rascher Spikes, registriert werden.
Insbesondere die Differentialdiagnose zum Pseudo-Lennox-Syndrom muss abgeklärt werden, welches sich dadurch vom LGS unterscheidet, dass keine tonischen Anfälle auftreten. Aufgrund dessen muss die Diagnose durch EEG im Schlaf (Schlaf-EEG) erfolgen, da die für das LGS typischen tonischen Anfälle meist im Schlaf auftreten.
Für die Überprüfung des Vorliegens einer hirnorganischen Besonderheit ist eine bildgebende Untersuchung des Gehirns mittels Magnetresonanztomographie (MRT) möglich. Treten die Bewegungsmuster bei insbesondere den tonischen Anfällen seitenbetont auf, lässt dies zum Beispiel eine Hirnschädigung der entsprechenden Seite vermuten, der nachgegangen werden sollte.
Bei der allgemeinmedizinischen Untersuchung fallen bei den meisten Kindern mit LGS im physischen Bereich Besonderheiten auf, insbesondere ist eine deutliche Entwicklungsverzögerung zu erkennen. Darüber hinaus zeigen sich vielfach kognitive Schwächen, eine entsprechende Einschränkung der Leistungsfähigkeit und Verhaltensauffälligkeiten. Die Besonderheiten können schon vor dem erstmaligen Auftreten der epileptischen Anfälle in Erscheinung getreten sein, aber auch erst nach bis zu zwei Jahren nach der Manifestation des LGS auftreten und sind vielfach auf die Grunderkrankung zurückzuführen, die zu den Anfällen geführt hat.
Therapie
Das LGS ist eine vergleichsweise schwer behandelbare Form von Epilepsie, wenngleich sie z. B. oft besser therapierbar ist als die Anfälle, die beim West-Syndrom auftreten. Eine möglichst frühzeitige Diagnose ist wichtig, kann jedoch einen Therapieerfolg nicht garantieren.
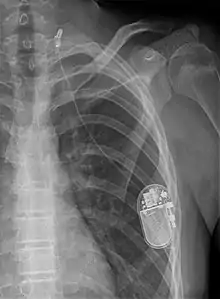
Ist eine behandelbare hirnorganische Besonderheit Ursache der Anfälle, ist in einigen Fällen nach gründlicher Abwägung der Vor- und Nachteile eine operative Korrektur durch Epilepsiechirurgie möglich, und durch die Beseitigung der Ursache können die Anfälle reduziert werden oder verschwinden. Treten (Sturz-)Anfälle sehr häufig auf und lassen sie sich medikamentös nicht einstellen, kann eine Vagusnervstimulation und ein teilweises Durchtrennen des Balkens, der die beiden Großhirnhälften verbindet, in Erwägung gezogen werden (Callosotomie).
In etwa einem von drei Fällen kann offenbar eine konsequente Ketogene Diät helfen, die gerade auch bei therapieresistenten Epilepsien eingesetzt wird.
Meist basiert die Therapie des LGS jedoch auf der Gabe von Medikamenten, wobei die medikamentöse Behandlung vergleichsweise schwierig ist und sich oft trotz aller Bemühungen keine Anfallsfreiheit erreichen lässt. Teils entwickelt sich eine Therapieresistenz oder eine Therapieresistenz liegt von Anfang an vor.
Ist eine Therapie mit nur einem Medikament (Monotherapie) nicht erfolgreich, werden mehrere, meist jedoch nicht mehr als drei, Medikamente gleichzeitig verabreicht (Kombinationstherapie). Häufig zum Einsatz kommen in der Behandlung des LGS die Arzneistoffe Valproat, Levetiracetam, Lamotrigin, Topiramat, Benzodiazepine und Felbamat. Mit dem Antiepileptikum Rufinamid steht eine neue Möglichkeit für die Zusatztherapie des LGS bei Patienten ab dem vierten Lebensjahr zur Verfügung; es kann zu einem signifikanten Rückgang der Anfallsfrequenz und des Schweregrads der Anfälle kommen. Seit 2018 ist das Medikament Epidiolex, welches zu 99,9 % aus Cannabidiol pflanzlichen Ursprungs besteht, in den USA zur Behandlung zugelassen.
Der Verlauf und die medikamentöse Behandelbarkeit sind beim Pseudo-Lennox-Syndrom in der Regel deutlich günstiger.
Prognose
Allgemein ist das LGS (bzw. die dem Epilepsie-Syndrom häufig zugrunde liegende Schädigung des Gehirns) durch eine vergleichsweise ungünstige Prognose gekennzeichnet – sowohl in Bezug auf die medizinische Behandelbarkeit als auch auf die Lebensqualität und die Kindesentwicklung, sowie in manchen Fällen auch auf die Lebensdauer. Unabhängig von Ausmaß und Auswirkung der Grunderkrankung werden ein früher Beginn des LGS und häufige tonische Anfälle als Merkmal einer ungünstigen Gesamtprognose gesehen. Zum Teil werden im Verlauf des LGS die epileptischen Anfälle zweitrangig, während oftmals fortschreitende kognitive und körperliche Beeinträchtigungen sowie Verhaltensstörungen in den Vordergrund treten.
Statistisch gesehen überleben fünf von 100 Kindern die ersten fünf Jahre ihres Lebens nicht, wobei sie nicht an den epileptischen Anfällen, sondern an der die Anfälle auslösenden Grunderkrankung oder daraus resultierenden Komplikationen sterben.
Statistisch gesehen können wenig mehr als die Hälfte der Kinder mit LGS durch Medikamente vergleichsweise befriedigend behandelt werden, wobei nur 5 bis 10 Kinder langfristig anfallsfrei bleiben und sich lediglich 15 von 100 Kindern kognitiv und motorisch regelgerecht entwickeln und im Erwachsenenalter ein selbständiges und weitgehend beschwerdefreies Leben führen. Letztere sind in aller Regel diejenigen, bei denen auch vor dem erstmaligen Auftreten des LGS keine Auffälligkeiten bestanden und keine Grunderkrankung (Hirnschädigung) als Ursache der Anfälle vorliegt.
Eine Zunahme von Entwicklungsauffälligkeiten ist in der Regel bei solchen Kindern zu beobachten, bei denen schon vor der Erstmanifestation des LGS Störungen der kognitiven und psycho-motorischen Entwicklung bestanden. Dieser Verlauf ist jedoch häufig nicht auf die epileptischen Anfälle, sondern auf die Grunderkrankung zurückzuführen, z. B. auf eine fortschreitende Fehlentwicklung des Gehirns. Aber auch ein ungünstiger Verlauf der epileptischen Anfälle, insbesondere bei statusartigem Auftreten der Krämpfe, kann zusätzliche bleibende Schädigungen verursachen.
Viele Kinder sind auch nach erfolgreicher Einstellung der Anfälle körperlich und kognitiv deutlich beeinträchtigt, wobei dies in der Regel nicht in erster Linie auf die epileptischen Anfälle, sondern auf deren Ursache (hirnorganische Besonderheit bzw. dessen Schweregrad) zurückgeführt werden kann. Häufig zeigen sich auch im Erwachsenenalter Lernstörungen, Sprachstörungen und Bewegungsstörungen sowie eine Einschränkung der kognitiven Leistungsfähigkeit und Cerebralparesen.
Siehe auch
Literatur
- Ulrich Altrup, Christian E. Elger: Epilepsie. Informationen in Texten und Bildern für Betroffene, Angehörige und Interessierte. Novartis Pharma-Verlag, Nürnberg 2000, ISBN 3-933185-49-1.
- Laura Doermer: Moritz mein Sohn. Bertelsmann, München 1990, ISBN 3-570-08222-9 (Erfahrungsbericht).
- Günter Krämer: Diagnose Epilepsie. Trias, Stuttgart 2003, ISBN 3-8304-3077-9.
- Günter Krämer, Ritva A. Sälke-Kellermann (Hrsg.): Das Lennox-Gastaut-Syndrom (= Epilepsie-Berichte. Band 5). Blackwell Wissenschafts-Verlag, Berlin u. a. 1998, ISBN 3-89412-341-9.
- Heiko Puckhaber: Epilepsie im Kindesalter. Eine interdisziplinäre Aufgabe. 5., unveränderte Auflage. Klotz, Eschborn bei Frankfurt am Main 2000, ISBN 3-88074-240-5.
- Hansjörg Schneble: Epilepsie bei Kindern. Wie Ihre Familie damit leben lernt. Trias, Stuttgart 1999, ISBN 3-89373-528-3.
- Ulrich Stephani: Das Lennox-Gastaut-Syndrom. Diagnose, Behandlung und Unterstützung im Alltag. Trias, Stuttgart 2008, ISBN 978-3-8304-3467-2.
Einzelnachweise
- W. G. Lennox, J. P. Davis: Clinical correlates of the fast and the slow spike-wave electroencephalogram. In: Pediatrics. Band 5, 1950, S. 626–644.
- H. Gastaut, H. Régis: On the subject of Lennox’ “akinetic” petit mal. In memory of W. G. Lennox. In: Epilepsia. Band 2, 1961, S. 298–305.
- H. Gastaut, J. Roger, R. Soulayrol u. a.: Childhood epileptic encephalopathy with diffuse slow spike-waves (otherwise known as ʻpetit mal variant’) or Lennox-syndrome. In: Epilepsia. Band 7, 1966, S. 139–179.
- C. Dravet: Encéphalopathie Épileptique de l’Enfant avec Pointe-onde lente diffuse (“petit mal variant”). Thesis. Marseille 1965.