Kontinuierliche Destillation
Die kontinuierliche Destillation ist eine Art der Destillation, die zu den Trennverfahren gehört. Bei der kontinuierlichen Destillation wird das Ausgangsgemisch kontinuierlich (d. h. ohne Unterbrechung) in das Verfahren eingebracht und auch die getrennten Fraktionen in kontinuierlichen Strömen ausgebracht.


Jede Fraktion kann eine oder mehrere Komponenten (Arten von chemischen Verbindungen) enthalten. Beim Destillieren von Erdöl oder einem ähnlichen Ausgangsmaterial enthält jede Fraktion viele Komponenten mit ähnlicher Flüchtigkeit und anderen Eigenschaften. Obwohl ein kontinuierlicher Destillationsbetrieb auch im kleinen Maßstab oder im Labor möglich ist, kommt die kontinuierliche Destillation am häufigsten in industriellen Großanlagen zum Einsatz.
Funktionsprinzip
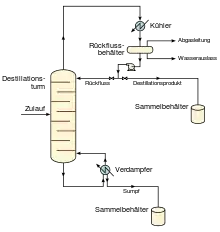
Grundlagen zu Destillation
Eine Destillation ist eine Trennung oder teilweise Trennung eines flüssigen Ausgangsgemischs in mehrere Komponenten bzw. Fraktionen durch selektives Verdampfen (Evaporation) und darauf folgendes Kondensieren aufgrund verschiedener Siedepunkte der Komponenten. Die Arbeitstemperatur liegt zwischen den Siedepunkten der zu trennenden Bestandteile. Es entstehen so mindestens zwei Fraktionen, von denen zumindest eine die flüchtige Destillat-Fraktion ist, die verdampft und dann gekühlt wird, wobei sie als wieder zu einer Flüssigkeit kondensierter Dampf aufgefangen wird. Die Bodenfraktion (die Restkomponente), nämlich der bei der Arbeitstemperatur nicht verdampfende Rückstand, verbleibt im Ausgangsbehälter.
Kontinuierliche Destillation
Das Funktionsprinzip bei einer kontinuierlichen Destillation unterscheidet sich nicht von dem einer normalen Destillation. Wenn ein flüssiges Gemisch zum Sieden gebracht wird, unterscheidet sich die Zusammensetzung des Dampfes über der Flüssigkeit von der Zusammensetzung der Flüssigkeit selbst. Wird dieser Dampf sodann getrennt und zu einer Flüssigkeit kondensiert, weist diese einen höheren Anteil der niedriger siedenden Komponente(n) des ursprünglichen Gemischs auf. Genau dieser Prozess läuft auch in einer Destillationskolonne ab, die für die kontinuierliche Destillation genutzt wird. Ein Gemisch wird erhitzt und in die Destillationskolonne geführt. Beim Eintritt in die Kolonne beginnt das Ausgangsmaterial nach unten abzufließen, aber ein Teil davon, nämlich die Komponente(n) mit dem niedrigeren Siedepunkt (mit den niedrigeren Siedepunkten), verdampft (verdampfen) und steigt (steigen) auf. Beim Aufsteigen kühlt der Dampf ab. Während nun ein Teil weiterhin als Dampf aufsteigt, beginnt der andere Teil (welcher einen höheren Anteil der weniger flüchtigen Komponente enthält) bereits, sich wieder nach unten zu bewegen.
Bild 3 zeigt eine einfache Fraktionierungskolonne, die für die kontinuierliche Destillation genutzt wird. In ihr wird der Ausgangsstrom in zwei Fraktionen getrennt, und zwar in ein Kopfprodukt und ein Bodenprodukt. Die „leichtesten“ Produkte (d. h. die Produkte mit dem niedrigsten Siedepunkt bzw. die flüchtigsten Produkte) treten aus der Oberseite der Kolonnen aus und die „schwersten“ Produkte (der Sumpf, d. h. die Produkte mit dem höchsten Siedepunkt) treten aus der Unterseite der Kolonne aus. Die Kopffraktion kann mit einem wassergekühlten oder luftgekühlten Kondensator gekühlt und kondensiert werden. Bei dem Sumpfverdampfer kann es sich um einen dampfbeheizten oder ölbeheizten Wärmetauscher oder sogar um einen Ofen mit Gas- oder Ölfeuerung handeln.
Bei einer kontinuierlichen Destillation wird das System in einem stationären Zustand oder einem annähernd stationären Zustand gehalten. Unter stationärer Zustand versteht man in diesem Kontext, dass sich die prozessrelevanten Mengen im Betrieb nicht im Laufe der Zeit ändern. Zu diesen konstanten Größen gehören die Menge des zugeführten Ausgangsmaterials, die Menge des abgegebenen Materialstroms, die Heizrate und die Kühlrate, das Rücklaufverhältnis sowie die Temperaturen, Drücke und Zusammensetzungen an jeder Stelle des Prozesses. Wenn der Prozess nicht durch Änderungen der Menge des zugeführten Gemischs, der zugeführten Wärmeenergie, der Umgebungstemperatur oder bezüglich der Kondensation gestört wird, bleibt der stationäre Zustand normalerweise aufrecht. Abgesehen von dem minimalen Kontrollaufwand (der im Übrigen unschwer durch Instrumente beherrschbar ist) ist dies der Hauptgrund, der die kontinuierliche Destillation so ausgesprochen attraktiv macht. Gelingt es, die Menge des zugeführten Ausgangsgemischs und die Zusammensetzung des Ausgangsgemischs konstant zu halten, so bleiben auch die Produktmenge und Produktqualität konstant. Selbst wenn es zu einer Änderung des Zustandes kommt, stehen im Normalfall moderne Prozesssteuerungsverfahren zur Verfügung, um das kontinuierliche Verfahren wieder in den stationären Zustand zurückzuführen.
Bei einer kontinuierlichen Destillation handelt es sich oft um eine fraktionierte Destillation; es kann sich um eine Vakuumdestillation oder eine Dampfdestillation handeln.
Vergleich mit der Chargendestillation
Eine Alternative zur kontinuierlichen Destillation ist die Chargendestillation, bei der das Gemisch zu Beginn der Destillation in die Destillieranlage gegeben wird, die Destillatfraktionen im Zuge der Destillation zeitlich nacheinander (eine nach der anderen) verdampft und kondensiert werden und die verbleibende Bodenfraktion zum Schluss entfernt wird. Da hier jede Destillatfraktion zu verschiedenen Zeitpunkten verdampft und kondensiert wird, genügt für eine Chargendestillation ein einziger Destillatablauf (Exit Point) – das Destillat kann einfach auf ein anderes Sammelgefäß, einen Fraktionsauffangbehälter, umgeschaltet werden. Die Chargendestillation kommt oft zum Einsatz, wenn kleinere Mengen destilliert werden. Bei einer kontinuierlichen Destillation durchläuft im Gegensatz zur Chargendestillation jeder Fraktionsstrom das Verfahren gleichzeitig. Daher wird für jede Fraktion auch ein eigener Destillatablauf benötigt. Gibt es mehrere Destillatfraktionen, so befindet sich in der Praxis jeder Destillatablauf auf einer verschiedenen Höhe entlang der Fraktionierkolonne. Die Bodenfraktion kann aus dem Boden der Destillierkolonne bzw. Anlage abgelassen werden, wird aber häufig aus einem an die Unterseite der Kolonne angeschlossenen Reboiler entnommen.
Da bei einer kontinuierlichen Destillationsanlage im Gegensatz zu einer Chargendestillation das Ausgangsgemisch in konstanter Menge und nicht etwa auf einmal zugeführt wird, wird für eine kontinuierliche Destillation auch kein großes Destillationsgefäß bzw. kein Behälter für eine Chargenfüllung benötigt. Das Gemisch kann also direkt in die Kolonne, in der die Trennung selbst stattfindet, eingeführt werden. Die Höhe der Zufuhröffnung an der Kolonne kann sich je nach den Gegebenheiten unterscheiden, und sie ist so ausgelegt, dass sie die optimalen Ergebnisse bietet.
Auslegung und Betrieb
Die Auslegung und der Betrieb einer Destillationskolonne hängen vom Ausgangsmaterial und von den Zielprodukten ab. Bei einem einfachen Ausgangsmaterial mit zwei Komponenten können analytische Verfahren wie das McCabe-Thiele-Verfahren[1][2][3] oder die Fenske-Gleichung[1] als Hilfe für die Auslegung dienen. Bei einem Ausgangsmaterial mit vielen verschiedenen Komponenten werden computerisierte Simulationsmodelle sowohl für die Auslegung sowie in weiterer Folge beim Betrieb der Kolonne verwendet. Anhand von Modellen können auch bereits installierte Kolonnen für die Destillation von Gemischen optimiert werden, für die sie ursprünglich nicht vorgesehen waren.
Eine für die kontinuierliche Destillation verwendete Kolonne muss im Betrieb genau kontrolliert werden, so dass Änderungen in der Zusammensetzung des Ausgangsmaterials, der Betriebstemperatur und der Produktzusammensetzung erkannt werden. Viele dieser Aufgaben werden mit fortschrittlichen Computersteuerungsgeräten bewerkstelligt.
Materialzufuhr in die Kolonne
Für die Zufuhr von Ausgangsmaterial in die Kolonne gibt es verschiedene Möglichkeiten. Wird das Material aus einer Quelle zugeführt, deren Druck höher als derjenige der Destillationskolonne ist, so kann es einfach über ein Rohr in die Kolonne eingebracht werden. Ansonsten wird das Ausgangsmaterial in die Kolonne gepumpt oder in diese komprimiert. Bei dem Ausgangsmaterial kann es sich um überhitzten Dampf, gesättigten Dampf, ein teilweise verdampftes Flüssigkeits-Dampf-Gemisch, eine gesättigte Flüssigkeit (d. h. die Flüssigkeit befindet sich bei dem in der Kolonne vorherrschenden Druck am Siedepunkt) oder eine unterkühlte Flüssigkeit handeln. Ist das Ausgangsmaterial eine Flüssigkeit, deren Druck den Kolonnendruck bei weitem übersteigt und fließt sie über ein Druckminderventil direkt vor der Kolonne, so kommt es zu einer sofortigen Ausdehnung und einer teilweisen Entspannungsverdampfung, so dass beim Eintritt in die Destillationskolonne ein Flüssigkeits-Dampf-Gemisch entsteht.
Rückfluss
Bei großen industriellen Fraktionierungskolonnen wird die Produkttrennungseffizienz der Produkte durch den Rückfluss gesteigert.[4][1] Als Rückfluss wird jener Anteil des kondensierten, flüssigen Kopfprodukts aus einer Destillationskolonne bezeichnet, der an den oberen Teil der Kolonne zurückgeführt wird. In der Kolonne sorgt die nach unten laufende Rückflussflüssigkeit für die Kühlung und teilweise Kondensation der aufsteigenden Brüden, wodurch die Effizienz der Destillationskolonne erhöht wird. Je mehr Rückfluss zur Verfügung steht, desto besser erfolgt die Trennung der niedriger siedenden von den höher siedenden Komponenten des Ausgangsmaterials in der Kolonne. Das Verhältnis der Rücklaufmenge zum Destillatabzug bezeichnet man als Rücklaufverhältnis.
Wird ein Gleichgewicht zwischen Erhitzung mit Reboiler unten an der Kolonne sowie Kühlung mit kondensiertem Rückfluss oben an der Kolonne erzielt, so bleibt in der Kolonne entlang der vertikalen Achse ein Temperaturgefälle (bzw. eine stufenweise Temperaturdifferenz) aufrechterhalten, was gute Voraussetzungen für die Fraktionierung des Ausgangsgemischs bietet. Rückflussströme in der Mitte der Kolonne werden als Rezirkulationen bzw. Pump-Arounds bezeichnet.
Durch eine Abänderung des Rückflusses (in Verbindung mit Abänderungen bei der Ausgangsmaterialzufuhr und der Produktentnahme) kann die Trennleistung einer kontinuierlichen Destillationskolonne im Betrieb ebenfalls verbessert werden (im Gegensatz hierzu würde ein Anbringen zusätzlicher Platten oder Böden bzw. ein Wechseln der Füllkörper mindestens eine ziemlich erhebliche Stillstandzeit mit sich bringen).
Erhöhung der Trennleistung
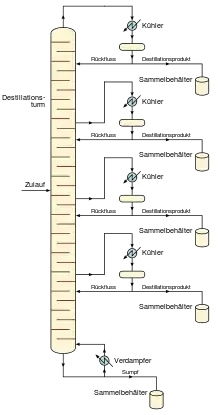
Obwohl kleine Einheiten, die im Allgemeinen aus Glas bestehen, in Labors verwendet werden können, kommen in industriellen Anlagen große, senkrechte, als „Destillationskolonnen“ bzw. „Trenntürme“ bezeichnete Stahlgefäße zum Einsatz. Zur Erhöhung der Trennleistung ist die Kolonne im Inneren normalerweise mit horizontalen Platten oder Böden ausgestattet, oder die Kolonne ist mit Füllkörpern gefüllt. Die bei der Destillation zur Verdampfung und zum Kompensieren von Wärmeverlusten erforderliche Wärme wird in den meisten Fällen von einem Reboiler an der Unterseite der Kolonne zugeführt. Die Reinheit des Kopfprodukts kann durch Rückführen eines Teils der extern kondensierten Kopfproduktflüssigkeit als Rücklauf verbessert werden. Abhängig von ihrem Zweck können an der vertikalen Achse der Destillationskolonnen in gewissen Abständen Flüssigkeitsauslässe vorhanden sein.
Aufbau der Kolonne
Platten oder Böden
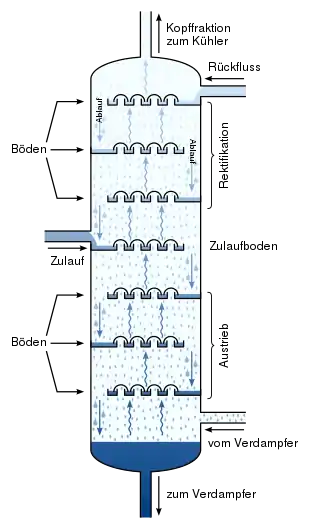
In Destillationskolonnen sorgen verschiedene Dampf- und Flüssigkeitskontaktverfahren für die erforderliche Anzahl an Gleichgewichtsstufen. Diese Elemente werden gewöhnlich als „Platten“ oder „Böden“ bezeichnet.[5] An jeder dieser Platten bzw. an jedem dieser Böden herrscht eine andere Temperatur und ein anderer Druck vor. Bei der Stufe am Kolonnenboden sind der Druck und die Temperatur am höchsten. Bewegen wir uns in der Kolonne Stufe um Stufe nach oben, so nehmen der Druck und die Temperatur jedes Mal ab. Das Dampf-Flüssigkeitsgleichgewicht für jede Ausgangsmaterialkomponente in der Kolonne reagiert spezifisch auf die verschiedenen Druck- und Temperaturbedingungen der jeweiligen Stufe. Dies bedeutet, dass jede Komponente in jeder Stufe eine bestimmte Konzentration in der Dampf- und Flüssigphase einstellt. Dies bewirkt die Trennung der Komponenten. Eine detailliertere, vergrößerte Abbildung von zwei Böden ist in dem Artikel theoretischer Boden zu sehen. Der Reboiler dient häufig als zusätzliche Gleichgewichtsstufe. Hätte jeder Boden bzw. jede Platte eine Effizienz von 100 %, dann entspräche die Anzahl der erforderlichen Böden, die für eine bestimmte Trennung benötigt würde, der Anzahl der Gleichgewichtsstufen bzw. theoretischen Platten. Dies ist allerdings nur sehr selten der Fall. Daher benötigt eine Destillationskolonne mehr Platten als die erforderliche Anzahl der theoretischen Dampf-Flüssigkeitsgleichgewichtsstufen.
Füllkörper
Zur Verbesserung der Trennung in einer Destillationskolonne kann jedoch auch Füllmaterial anstelle von Böden verwendet werden. Diese bieten den Vorteil eines geringeren Druckabfalls in der Kolonne (dies im Vergleich zu Platten oder Böden), was bei einem Betrieb im Vakuum von Vorteil ist. Kommt in einer Destillationskolonne Füllmaterial anstelle von Böden zum Einsatz, wird zunächst die Anzahl der erforderlichen theoretischen Gleichgewichtsstufen bestimmt. Dann wird die Füllkörperhöhe, welche einer theoretischen Gleichgewichtsstufe entspricht – diese wird als Trennstufenhöhe (height equivalent to a theoretical plate bzw. HETP) bezeichnet – ebenfalls ermittelt. Die insgesamt erforderliche Füllkörperhöhe entspricht der Anzahl der theoretischen Stufen multipliziert mit der Trennstufenhöhe.
Bei diesem Füllmaterial kann es sich entweder um eine Füllkörperschüttung wie z. B. Raschigringe oder aber um strukturiertes Blech handeln. Die Flüssigkeiten neigen dazu, die Oberfläche des Füllkörpers zu benetzen, und die Brüden ziehen über diese benetzte Oberfläche, an der ein Stoffaustausch stattfindet. Im Gegensatz zur herkömmlichen Bodendestillation, wo jeder Boden einem separaten Punkt des Dampf-Flüssigkeitsgleichgewichts entspricht, weist die Dampf-Flüssigkeitsgleichgewichtskurve in einer gefüllten Kolonne einen kontinuierlichen Verlauf auf. Bei der modellhaften Erfassung von gefüllten Kolonnen ist es jedoch nützlich, die Anzahl der theoretischen Böden zu berechnen, um den Trennwirkungsgrad der gefüllten Kolonne bezogen auf traditionellere Böden angeben zu können. Bei verschieden geformten Füllungen unterscheiden sich die Oberflächen und der leere Raum zwischen den Füllungen. Beide Faktoren beeinflussen die Leistung der Füllung.
Zusätzlich zur Füllungsform und Oberfläche ist auch die Verteilung von in das gefüllte Bett eintretender Flüssigkeit und Dampf ein weiterer Faktor, welcher die Leistung von Füllkörperschüttungen oder strukturierten Füllkörpern beeinflusst. Die Anzahl der theoretischen Stufen, die für die Durchführung einer bestimmten Trennung erforderlich sind, wird anhand eines spezifischen Dampf-Flüssigkeitsverhältnisses berechnet. Wenn die Flüssigkeit und der Dampf beim Eintritt in das gefüllte Bett nicht gleichmäßig über die Kolonnenoberfläche verteilt sind, stimmt das Dampf-Flüssigkeitsverhältnis im gefüllten Bett nicht, und die erforderliche Trennung wird nicht erzielt; die Füllung scheint nicht korrekt zu funktionieren und die Trennstufenhöhe (height equivalent to a theoretical plate bzw. HETP) ist größer als erwartet. Das Problem liegt aber nicht in der Füllung selbst, sondern in der schlechten Verteilung der in das gefüllte Bett eintretenden Flüssigkeiten. Häufiger liegt das Problem in einer Fehlverteilung der Flüssigkeit als des Dampfes. Das Design der zur Einleitung des Ausgangsmaterials und des Rückflusses in das gefüllte Bett zum Einsatz kommenden Flüssigkeitsverteiler ist ausschlaggebend dafür, ob die Füllung den maximalem Wirkungsgrad erreicht. In den Referenzen sind Verfahren zur Beurteilung der Wirksamkeit eines Flüssigkeitsverteilers zu finden.[6][7]
Kopfsystem
In Bild 4 und 5 wird davon ausgegangen, dass eine Kopffraktion mittels Wasser- oder Luftkühlung vollständig zu einer Flüssigkeit kondensiert wird. In vielen Fällen ist es jedoch schwierig, den Kolonnenkopf vollständig zu kondensieren. Dann muss der Rückflussbehälter über einen Gasauslass verfügen. In wieder anderen Fällen enthält die Kopffraktion unter Umständen Wasserdampf, entweder weil das zugeführte Ausgangsmaterial etwas Wasser enthält oder weil etwas Dampf in die Destillationskolonne eingedüst wird (wie zum Beispiel bei Erdöldestillationskolonnen in Ölraffinerien). Wenn das Destillatprodukt in Wasser unlöslich ist, befindet sich im Rückflussbehälter unter Umständen eine kondensierte Flüssigdestillatphase, eine kondensierte Wasserphase und eine nicht kondensierfähige Gasphase. Dann muss der Rückflussbehälter zusätzlich mit einem Wasserauslass ausgestattet sein.
Industrielle Nutzung
Allgemein
Die Destillation ist eine der Grundoperationen in der Verfahrenstechnik.[8][9] In chemischen Verfahrensindustrien wird die kontinuierliche Destillation insbesondere dann umfassend genutzt, wenn große Mengen an Flüssigkeiten destilliert werden müssen.[4][10][1] Beispiele hierfür sind die Erdgasverarbeitung, die Erzeugung von Petrochemikalien, die Teerverarbeitung, das Bierbrauen, die Trennung von verflüssigter Luft, die Erzeugung von Kohlenwasserstofflösemitteln und ähnliche Industrien, am intensivsten wird sie jedoch in Erdölraffinerien genutzt. Mit Erdöl kommt in diesen Raffinerien ein hochkomplexes Gemisch mit vielen verschiedenen, zu trennenden Komponenten als Ausgangsmaterial zum Einsatz. Das Ergebnis dieses Prozesses ist nicht die Trennung in reine chemischen Verbindungen, sondern lediglich die Trennung in Gruppen von Komponenten mit einem relativ engen Siedepunktbereich, die als Fraktionen bezeichnet werden. Von diesem Begriff „Fraktionen“ leitet sich auch die Bezeichnung fraktionierte Destillation bzw. Fraktionierung ab. In Anbetracht der Produktanforderungen und nach dem Gesichtspunkt der Wirtschaftlichkeit ist es oft nicht lohnend, eine weitere Trennung der Komponenten innerhalb dieser Fraktionen vorzunehmen.
Für die industrielle Destillation werden normalerweise große, senkrechte, zylinderförmige Kolonnen verwendet, die als „Trenntürme“ bzw. „Destillationskolonnen“ bezeichnet werden. Diese Kolonnen haben einen Durchmesser von etwa 65 Zentimeter bis 11 Meter und eine Höhe von etwa 6 bis 60 Meter und mehr.
Erdöl
In rohem Erdöl sind etliche hundert verschiedene Kohlenwasserstoffverbindungen enthalten: Paraffine, Naphthene und Aromaten sowie organische Schwefelverbindungen, organische Stickstoffverbindungen und einige sauerstoffhaltige Kohlenwasserstoffe wie zum Beispiel Phenole. Obwohl Rohöle normalerweise keine Olefine enthalten, werden sie in vielen Verfahren gebildet, die in einer Erdölraffinerie zum Einsatz kommen.[11]
Im Erdölfraktionierer entstehen keine Produkte, die einen einzigen Siedepunkt haben, sondern Fraktionen mit Siedebereichen.[11][12] So produziert der Erdölfraktionierer zum Beispiel eine als „Naphtha“ bezeichnete Kopffraktion, die nach weiterer Verarbeitung durch einen katalytischen Wasserstoffentschwefler zur Entfernung von Schwefel und einen katalytischen Reformer zur Umbildung seiner Kohlewasserstoffmoleküle in komplexere Moleküle mit höherer Oktanzahl ein Bestandteil von Benzin wird.
Die sogenannte Naphtha-Fraktion enthält viele verschiedene Kohlenwasserstoffverbindungen. Daher weist sie einen ersten Siedepunkt von etwa 35 °C und einen letzten Siedepunkt von etwa 200 °C auf. Jede in den Fraktionierkolonnen herausgebildete Fraktion hat einen anderen Siedebereich. Unter der Kopffraktion wird in einem bestimmten Abstand die nächste Fraktion seitlich aus der Kolonne entnommen – normalerweise handelt es sich dabei um die Düsenkraftstofffraktion, die auch als Kerosinfraktion bezeichnet wird. Der Siedebereich dieser Fraktion reicht von einem ersten Siedepunkt von etwa 150 °C bis zu einem letzten Siedepunkt von etwa 270 °C, und auch sie enthält viele verschiedene Kohlenwasserstoffe. Die nächste Fraktion in der Kolonne (in absteigender Richtung) ist die Dieselölfraktion mit einem Siedebereich von 180 °C bis etwa 315 °C. Der Siedebereich zwischen einer Fraktion und der nächsten Fraktion überlappen sich, da die Trennungen bei der Destillation keine perfekt scharf gezogene Linie darstellen. Die nächste Fraktion ist die Schwerölfraktion, und schließlich kommt noch die Bodenfraktion, deren Siedebereiche sehr breit sind. All diese Fraktionen werden in nachgelagerten Raffinerieprozessen weiteren Bearbeitungsschritten unterzogen.
Einzelnachweise
- Perry, Robert H. and Green, Don W.: Perry's Chemical Engineers' Handbook, 6th. Auflage, McGraw-Hill, 1984, ISBN 0-07-049479-7.
- Milton Beychok: Algebraic Solution of McCabe-Thiele Diagram. In: Chemical Engineering Progress. Mai 1951.
- Seader, J. D., und Henley, Ernest J.: Separation Process Principles. Wiley, New York 1998, ISBN 0-471-58626-9.
- Kister, Henry Z.: Distillation Design, 1st. Auflage, McGraw-Hill, 1992, ISBN 0-07-034909-6.
- Photographs of bubble cap and other tray types (Website of Raschig GmbH).
- Random Packing, Vapor and Liquid Distribution: Liquid and gas distribution in commercial packed towers, Moore, F., Rukovena, F., Chemical Plants & Processing, Edition Europe, August 1987, S. 11–15.
- Structured Packing, Liquid Distribution: A new method to assess liquid distributor quality, Spiegel, L., Chemical Engineering and Processing 45 (2006), 1011-1017.
- Editors: Jacqueline I. Kroschwitz und Arza Seidel: Kirk-Othmer Encyclopedia of Chemical Technology, 5th. Auflage, Wiley-Interscience, Hoboken, NJ 2004, ISBN 0-471-48810-0.
- McCabe, W., Smith, J. und Harriott, P.: Unit Operations of Chemical Engineering, 7th. Auflage, McGraw Hill, 2004, ISBN 0-07-284823-5.
- King, C.J.: Separation Processes, 2nd. Auflage, McGraw Hill, 1980, ISBN 0-07-034612-7.
- Gary, J.H. and Handwerk, G.E.: Petroleum Refining Technology and Economics, 2nd. Auflage, Marcel Dekker, Inc., 1984, ISBN 0-8247-7150-8.
- Nelson, W.L.: Petroleum Refinery Engineering, 4th. Auflage, McGraw Hill, 1958, LCCN 57010913.
Weblinks
- Distillation Theory (PDF; 184 kB) by Ivar J. Halvorsen and Sigurd Skogestad, Norwegian University of Science and Technology, Norway.
- Distillation, An Introduction by Ming Tham, Newcastle University, UK.
- Distillation by the Distillation Group, USA.
- Distillation Lecture Notes by Prof. Randall M. Price at Christian Brothers University.
- Petroleum Distillation by Wayne Pafco.
- Distillation simulation software.