Greig-Syndrom
Das Greig-Syndrom ist eine angeborene Erkrankung mit einer charakteristischen Kombination von Schädel-Gesichtsdysmorphie und Polydaktylie (Vielgliedrigkeit der Finger).[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.0 | Angeborene Fehlbildungssyndrome mit vorwiegender Beteiligung des Gesichtes |
| ICD-10 online (WHO-Version 2019) | |

Synonyme sind: Greig-Zephalopolysyndaktylie-Syndrom; Zephalopolysyndaktylie; Hootnick-Holmes-Syndrom
Nicht zu verwechseln ist der Familiäre Hypertelorismus Greig.[3][1]
Die Bezeichnungen beziehen sich auf den Autor der Erstbeschreibung 1926 durch den schottischen Arzt David Middleton Greig (1864–1936)[4][5] sowie die Autoren einer Publikation von 1972, den US-amerikanischen Orthopäden David Randall Hootnick und den US-amerikanischen Kinderarzt Levis B. Holmes.[6][7]
Vorkommen
Die Häufigkeit wird mit 1–9 zu 1.000.000 angegeben, die Vererbung erfolgt autosomal-dominant.[2]
Ursache
Der Erkrankung liegen ursächlich Loss-of-Function-Mutationen im GLI3-Gen auf Chromosom 7 am Genort p13 zugrunde, welches für einen Transkriptionsfaktor kodiert.[8]
Andere Mutationen im gleichen Gen finden sich bei dem Pallister-Hall-Syndrom und dem Akrokallosalen Syndrom.[2]
Klinische Erscheinungen
Diagnostische Kriterien sind:[1][2]
- Makrozephalus, prominente Stirn, später Fontanellenschluss
- Hypertelorismus
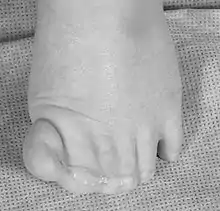
- Polydaktylie der Hände (postaxial) und der Füße (präaxial)
- Partielle häutige Syndaktylien der Finger & Zehen II-V
selten zusätzlich Balkenmangel, andere Anomalien des Gehirnes.
Diagnostik
An das Syndrom sollte beim Vorliegen der „klassischen“ Trias von präaxialer Polydaktylie mit häutiger Syndaktylie, Hypertelorismus und Makrozephalie gedacht werden.[2]
Differentialdiagnose
Heilungsaussicht
Es besteht ein etwas höheres Risiko einer auch geistig verzögerten Entwicklung.[2]
Literatur
- J. Mücke, K. R. Sandig: Greig-Syndrom. In: Kinderärztliche Praxis. Bd. 53, Nr. 2, Februar 1985, S. 89–91, ISSN 0023-1495. PMID 3921750.
- L. G. Biesecker: The Greig cephalopolysyndactyly syndrome. In: Orphanet Journal of Rare Diseases. Bd. 3, 2008, S. 10, ISSN 1750-1172. doi:10.1186/1750-1172-3-10. PMID 18435847. PMC 2397380 (freier Volltext). (Review).
Einzelnachweise
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Greig-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- D. M. Greig: Hypertelorism. A hitherto undifferentiated congenital cranio-facial deformity. In: Edinburgh Medical Journal, 1924, Bd. 31, S. 560–593.
- Who named it Greig
- D. M. Greig: Oxycephaly. Edinburgh Medical Journal, 1926, Bd. 33, S. 189–218.
- D. Hootnick, L. B. Holmes: Familial polysyndactyly and craniofacial anomalies. In: Clinical Genetics, Copenhagen, 1972, Bd. 3, S. 128–134.
- Who named it Greig's Syndrom
- Greig cephalopolysyndactyly syndrome. In: Online Mendelian Inheritance in Man. (englisch)
Weblinks
- L. G. Biesecker und J. J. Johnston: Greig Cephalopolysyndactyly Syndrome. In: GeneReviews®
- Medline Plus