Akrokallosales Syndrom
Akrokallosales Syndrom ist eine sehr seltene angeborene Erkrankung mit den Hauptmerkmalen Agenesie des Corpus callosum (CC), distal betonte Fehlbildungen der Extremitäten, insbesondere Polydaktylie, Gesichtsauffälligkeiten und geistige Behinderung.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q04.0 | Angeborene Fehlbildungen des Corpus callosum Inkl.: Agenesie des Corpus callosum |
| ICD-10 online (WHO-Version 2019) | |
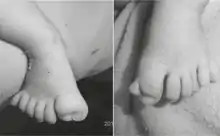
Synonyme sind: Acrocallosal-Syndrom; ACLS; ACS Schinzel-Typ; englisch Hallux-duplication; Absence of Corpus Callosum, Schinzel Type; Hallux Duplication, Postaxial Polydactyly, and Absence of Corpus Callosum
Der Erstbeschrieb erfolgte 1979 durch den österreichischen Humangenetiker Albert Schinzel[3][4].
Vorkommen
Die Häufigkeit (Prävalenz) wird mit unter 1 zu 1 Million angegeben, bislang wurde über etwa 50 Betroffene berichtet. die Vererbung erfolgt autosomal-rezessiv.[2]
Ursache
Der Erkrankung liegen entweder Mutationen im KIF7-Gen auf Chromosom 15 Genort q26.1, welches für das kinesin family member 7 kodiert, oder im GLI3-Gen auf Chromosom 7 an p14.1 zugrunde, das für einen Transkriptionsfaktor kodiert.[5]
Beide Gene sind im Sonic Hedgehog-Signalweg beteiligt.[2]
Mutationen im KIF7-Gen finden sich auch im Joubert-Syndrom Typ 12.[6]
Andere Mutationen im GLI3-Gen finden sich beim Greig-Syndrom.
Klinische Erscheinungen
Klinische Kriterien sind:[1][2]
- Balkenmangel
- Arachnoidalzyste (1/3)
- Unterentwicklung von Hirnabschnitten (Medulla oblongata, Temporallappen, Pons oder des Kleinhirnwurmes)
- Mikropolygyrie
- Makrozephalie mit prominenter Stirn, betontes Hinterhaupt, großer vorderer Fontanelle
- Hypertelorismus, antimongoloide Lidstellung
- kurzer Unterkiefer
- nach vorne gerichtete Nasenlöcher und breiter flacher Nasenrücken
- Klinodaktylie des V. Fingers, Polydaktylie oder Syndaktylie von Zehen und Finger
Seltener treten auf: kurzes Philtrum/Oberlippe, hoher Gaumenbogen, Lippen-/Gaumenspalte, Herzfehler, Hypospadie und Hernien.
Diagnostik
Eine vorgeburtliche Diagnostik ist ab der 20. Schwangerschaftswoche durch Ultraschall und Magnetresonanz-Bildgebung möglich[2].
Differential-Diagnose
- Zephalo-Polysyndaktylie Greig
- Oro-fazio-digitales Syndrom Typ 1 und Typ 2
- Meckel-Gruber-Syndrom
- Smith-Lemli-Opitz-Syndrom
- Rubinstein-Taybi-Syndrom
- Pena-Shokeir-Syndrom Typ 2 (Zerebro-okulo-fazio-skeletales Syndrom)
- Aicardi-Syndrom
- Neu-Laxova-Syndrom
- Pseudotrisomie-13-Syndrom
- Toriello-Carey-Syndrom
- Oto-palato-digitales Syndrom Typ 2
- Da Silva-Syndrom[2]
Therapie
Die Prognose wird durch den Schweregrad der Fehlbildungen, durch die Muskelhypotonie und durch das Auftreten von Krampfanfällen bestimmt.[2]
Einzelnachweise
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Akrokallosal-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- whonamedit
- A. Schinzel: Postaxial polydactyly, hallux duplication, absence of the corpus callosum, macrencephaly and severe mental retardation: a new syndrome? In: Helvetica paediatrica acta. Band 34, Nummer 2, Mai 1979, S. 141–146, ISSN 0018-022X. PMID 457430.
- D. M. Walsh, S. A. Shalev, M. A. Simpson, N. V. Morgan, Z. Gelman-Kohan, J. Chemke, R. C. Trembath, E. R. Maher: Acrocallosal syndrome: identification of a novel KIF7 mutation and evidence for oligogenic inheritance. In: European journal of medical genetics. Band 56, Nummer 1, Januar 2013, S. 39–42, ISSN 1878-0849. doi:10.1016/j.ejmg.2012.10.004. PMID 23142271.
- Acrocallosal syndrome. In: Online Mendelian Inheritance in Man. (englisch)
Literatur
- B. Hosdurg, A. P. Nair, J. Simha, S. Sriramanan: Anaesthetising an infant with acrocallosal syndrome: An unusual case. In: Indian journal of anaesthesia. Band 62, Nummer 5, Mai 2018, S. 389–391, doi:10.4103/ija.IJA_449_17, PMID 29910499, PMC 5971630 (freier Volltext).
(26648833)
- R. Singhal, S. Pandit, A. Saini, P. Singh, N. Dhawan: The acrocallosal syndrome in a neonate with further widening of phenotypic expression. In: Iranian journal of child neurology. Band 8, Nummer 2, 2014, S. 60–64, PMID 24949054, PMC 4058068 (freier Volltext).
- V. V. Ramteke, P. A. Darole, Z. F. Shaikh, N. J. Padwal, B. Agrawal, M. S. Shrivastava, S. Kamath: Acrocallosal syndrome in a young hypertensive male. In: BMJ Case Reports. Band 2011, Mai 2011, S. , doi:10.1136/bcr.12.2010.3648, PMID 22696705, PMC 3089937 (freier Volltext).
- R. Koenig, A. Bach, U. Woelki, K. H. Grzeschik, S. Fuchs: Spectrum of the acrocallosal syndrome. In: American journal of medical genetics. Band 108, Nummer 1, Februar 2002, S. 7–11, ISSN 0148-7299. PMID 11857542.
- A. Schinzel: The acrocallosal syndrome in first cousins: widening of the spectrum of clinical features and further support for autosomal recessive inheritance. In: Journal of medical genetics. Band 25, Nummer 5, Mai 1988, S. 332–336, ISSN 0022-2593. PMID 3385741. PMC 1050460 (freier Volltext).