1q21.1-Deletionssyndrom
Das 1q21.1-Deletionssyndrom ist ein seltenes Syndrom, welches durch eine Deletion auf dem menschlichen Chromosom 1 an der Stelle 1q21.1 verursacht wird. Folgen dieser Veränderung können mentale Retardierung und verschiedene körperliche Anomalien sein. Die Penetranz und Expressivität sind variabel. Einige Menschen mit dieser Deletion weisen keine erkennbare Beeinträchtigung auf.
| Klassifikation nach ICD-10 | |
|---|---|
| Q93.5 | Sonstige Deletionen eines Chromosomenteils |
| ICD-10 online (WHO-Version 2019) | |
Unique, eine internationale Vereinigung von Menschen mit seltenen chromosomalen Abweichungen, kennt 64 registrierte Menschen mit dieser Deletion weltweit. (Stand: Oktober 2012)[1]
Neben der 1q21.1-Deletion gibt es auch eine 1q21.1-Duplikation, bei der der betreffende Abschnitt zwei- oder dreifach vorhanden ist.
Struktur der Region 1q21.1
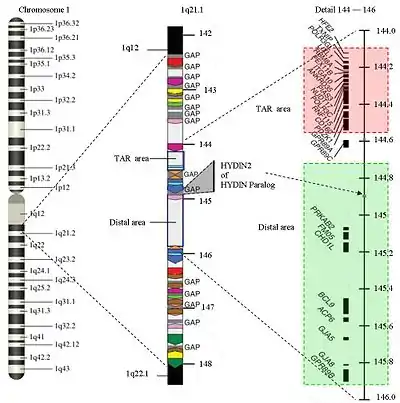
Die Struktur der 1q21.1 ist komplex. Das Gebiet hat eine Größe von ca. 6 Millionen Basenpaaren (Mb) (von 141,5 auf 147,9 Mb). Es gibt zwei Bereiche, in denen die Deletion auftreten kann: den TAR-Bereich mit der Folge eines TAR-Syndroms und den distalen Bereich, der zu anderen Anomalien führt. Das Gebiet weist mehrere Wiederholungen der gleichen Struktur auf (Bereiche in der gleichen Farbe auf dem Bild haben gleiche Strukturen). Nur 25 % der Struktur sind spezifisch. Bis heute gibt es jedoch keine vollständigen Informationen über die Nukleotidsequenz in diesen Bereichen. Der Bereich 1q21.1 gilt als einer der schwierigsten bei der Kartierung des menschlichen Genoms. Die fehlenden Bereiche betragen derzeit etwa 700.000 Basenpaare. Daher ist es schwer, Beginn und Ende einer Deletion genau zu bestimmen.
Typen
Man unterscheidet beim 1q21.1-Deletionssyndrom zwei Typen:
- Die sogenannte Klasse-I-Deletion ist auf den TAR-Bereich oder den distalen Bereich beschränkt.
- Ist die Deletion so groß, dass beide Bereiche betroffen sind, wird sie als Klasse-II-Deletion bezeichnet. Dabei gibt es komplexe Fälle, in denen sowohl der TAR- als auch der distale Bereich betroffen sind, während der dazwischenliegende Bereich normal ist, sowie auch einige atypische Varianten.
Eine normale Deletion betrifft zwischen 1,0 und 1,9 Millionen Basenpaare (Mb). Nach Mefford sind 1,35 Mb der Standard für eine solche Deletion[2]. Die größte am lebenden Menschen beobachtete Deletion beträgt über 5 Mb.
Symptome
Die Folgen der Mikrodeletion sind phänotypisch sehr variabel. Während manche Menschen mit diesem Syndrom keinerlei erkennbare Beeinträchtigungen aufweisen, kommt es bei anderen zu erheblichen Einschränkungen. Bislang nachgewiesene Symptome sind:
- Haploinsuffizienz
- TAR-Syndrom
- neurologisch-psychiatrische Probleme: Entwicklungsretardierung, mentale Retardierung, Autismus, Schizophrenie[3][4], Epilepsie, ataktischer Gang mit Sturzneigung
- Dysmorphien: Mikrozephalie, vorstehende Stirn, Knollennase, breite Daumen und Zehen, zusätzliche Querfalte des fünften Fingers (Klasse-II-Deletion), Hypermobilität der Gelenke, Pseudarthrose des Schlüsselbeines (Klasse-II-Deletion)[5], Vaginalaplasie („Müllersche Aplasie“)
- Herz-Anomalien und kardiovaskuläre Anomalien (30 % der Fälle) wie ein anomaler Ursprung der Koronargefäße (Klasse-II-Deletion)
- ophthalmologische Probleme: Grauer Star, tiefliegende Augen, Schielen
- Nierenerkrankungen: fehlende Niere, Wanderniere, Neuroblastom[6]
Folgende Symptome konnten bisher nicht sicher dem 1q21.1-Deletionssyndrom zugeordnet werden:
- Rückfluss von Magensäure in die Speiseröhre
- Non-Compaction-Kardiomyopathie in Zusammenhang mit einem Klasse-II-1q21.1 Deletionssyndrom.[7]
- erhöhte Nackentransparenz und Oligohydramnion während einer Schwangerschaft[8].
Da nur wenige Informationen über das Syndrom vorliegen, ist die Vollständigkeit obiger Symptomliste unsicher.
Betroffene Gene
Die beim 1q21.1-Deletionssyndrom im TAR-Bereich betroffenen Gene sind: HFE2 (Hämojuvelin), TXNIP, POLR3GL, LIX1L, RBM8A, PEX11B, ITGA10, ANKRD35, PIAS3, NUDT17, POLR3C, RNF115, CD160, PDZK1 und GPR89A.
Die im distalen Bereich betroffenen Gene sind PDE4DIP, HYDIN2, PRKAB2, PDIA3P, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8, NBPF10, GPR89B, GPR89C, PDZK1P1 und NBPF11.
Diagnostik
Das Syndrom kann auch in Familien, in denen keiner der Elternteile die Gene trägt, auftreten. Wegen der Wiederholungen in 1q21.1 besteht eine höhere Wahrscheinlichkeit für ein ungleiches Crossing-over während der Meiose. In diesem Fall können Teile des Chromosoms verloren gehen, und es kommt zu Kopienzahlvariationen (Copy Number Variation – CNV) in Form von Deletionen oder Duplizierungen. So eine zufällige Mutation wird eine „de-novo“-Situation genannt und tritt in 75 % der Fälle auf.
In 25 % der Fälle ist einer der Elternteile Träger des Syndroms, ohne dass es sich auf die Person auswirkt, oder es bestehen nur leichte Symptome. In mehreren Fällen wurde das Syndrom zunächst bei Kindern wegen Autismus oder eines anderen Problems diagnostiziert und erst später stellte sich heraus, dass auch einer der Elternteile betroffen war.
In Familien, in denen beide Eltern negativ auf das Syndrom getestet wurden, ist das Risiko, dass ein zweites Kind mit dem Syndrom zur Welt kommt, äußerst gering. Wenn das Syndrom in der Familie gefunden wurde, besteht ein Erkrankungsrisiko von 50 %, weil ein autosomal dominanter Erbgang vorliegt. Die Auswirkungen der 1q21.1-Mikrodeletion auf das Kind können jedoch nicht vorhergesagt werden. Eltern, deren Kind am Syndrom erkrankt ist, sollten daher vor einer weiteren Schwangerschaft humangenetisch beraten und untersucht werden.
Eine Feststellung der Chromosomveränderung ist durch Fluoreszenz-in-situ-Hybridisierung (FISH) möglich. In der Schwangerschaft ist im Rahmen der Pränataldiagnostik eine Feststellung ebenfalls möglich.
Therapie
Aufgrund der genetischen Ursache ist eine ursächliche Behandlung nicht möglich. Angezeigt ist jedoch eine symptomatische Therapie, insbesondere die Korrektur der auftretenden Fehlbildungen und die Therapie der Begleiterkrankungen.
Forschung
Das Syndrom wurde erstmals bei Menschen mit Herzabnormalitäten diagnostiziert, später jedoch auch bei Patienten gefunden, die an Autismus und Schizophrenie litten. Aufgrund wissenschaftlicher Untersuchungen wurde deutlich, dass 20 von 1000 Patienten mit Autismus eine 1q21.1-Mikrodeletion haben.
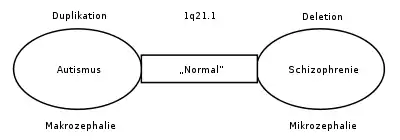
Das 1q21.1-Deletionssyndrom wird derzeit an mehreren Standorten weltweit untersucht. Dabei gibt es Hinweise auf einen Zusammenhang zwischen Autismus und Schizophrenie, der durch Duplizierungen und Deletionen an Chromosomen seinen Ursprung in der Embryogenese haben könnte. Statistische Untersuchungen zeigten, dass Schizophrenie bei einer 1q21.1-Mikrodeletion signifikant häufiger ist. Dagegen ist Autismus bei einer 1q21.1-Mikroduplizierung signifikant häufiger. Ähnliche Beobachtungen wurden im Hinblick auf Chromosom 16 auf 16p11.2 (Deletion: Autismus / Duplizierung: Schizophrenie) sowie auf Chromosom 22 (22q11.21-Deletion (Velo-Cardio-Facial Syndrom): Schizophrenie / Duplizierung: Autismus) und 22q13.3 (Deletion (Phelan-McDermid-Syndrom): Schizophrenie / Duplizierung: Autismus) gemacht. Weitere Untersuchungen bestätigen eine mindestens 7,5%ige Wahrscheinlichkeit für einen Zusammenhang zwischen Schizophrenie und Deletionen auf 1q21.1, 3q29, 15q13.3, 22q11.21 en Neurexin 1 (NRXN1) und Duplizierung auf 16p11.2.[9][10]
Untersuchungen zu den Beziehungen zwischen Autismus, Schizophrenie und Mikroveränderungen am Chromosom 15 (15q13.3) sowie am Chromosom 16 (16p13.1) und Chromosom 17 (17p12) sind noch nicht schlüssig (Stand 2011).
Variationen des BCL9-Gens, das sich im distalen Bereich befindet, verstärken das Risiko auf Schizophrenie und führen möglicherweise zu bipolaren Störungen und Depressionen.[11]
Die Forschung konzentriert sich derzeit auf zehn bis zwölf Gene auf 1q21.1, welche für die Produktion von DUF1220 verantwortlich sind. DUF1220 ist eine Proteindomäne unbekannter Funktion, die in den Neuronen des Gehirns in der Nähe des Neocortex aktiv ist. Basierend auf Forschungen an Affen und anderen Säugetieren wird davon ausgegangen, dass die Anzahl der DUF1220-Genloci mit der kognitiven Entwicklung in Beziehung steht (Mensch: 212 ; Schimpanse: 37; Affen: 30; Maus: 1). Es scheint, dass die DUF1220-Orte auf 1q21.1 sich an Stellen befinden, die mit der Größe und der Entwicklung des Gehirns zusammenhängen, was wiederum mit Autismus (Makrozephalie) und Schizophrenie (Mikrozephalie) verbunden ist. Es wird davon ausgegangen, dass eine Deletion oder eine Duplizierung eines Gens, das DUF1220-Gebiete hervorbringt, Wachstums- und Entwicklungsstörungen des Gehirns verursachen kann.
In der Erforschung der HYDIN2- oder HYDIN-Paralogie ist ein weiterer Zusammenhang zwischen Makrozephalie und Duplizierungen sowie zwischen Mikrozephalie und Deletionen entdeckt worden. Dieser Teil des 1q21.1 ist an der Entwicklung des Gehirns beteiligt. Es wird angenommen, dass es ein dosisempfindliches Gen ist. Wenn dieses Gen im 1q21.1-Bereich nicht zur Verfügung steht, führt es zu Mikrozephalie. Das HYDIN2 ist eine Kopie des HYDIN, das auf 16q22.2 gefunden wurde.
Quellen
- Jane Gregory: Bringing up a challenging child at home : when love is not enoug. Jessica Kingsley Publishers, London/ Philadelphia, PA 2000, ISBN 1-85302-874-6.
- Samantha J. L. Knight (Hrsg.): Genetics of mental retardation : an overview encompassing learning disability and intellectual disabilit. Karger, Basel/ New York 2010, ISBN 978-3-8055-9280-2.
- H. C. Mefford, A. J. Sharp, C. Baker u. a.: Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. In: N. Engl. J. Med. Band 359, Nr. 16, Oktober 2008, S. 1685–1699, doi:10.1056/NEJMoa0805384, PMID 18784092, PMC 2703742 (freier Volltext).
- N. Brunetti-Pierri, J. S. Berg, F. Scaglia u. a.: Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. In: Nat. Genet. Band 40, Nr. 12, Dezember 2008, S. 1466–1471, doi:10.1038/ng.279, PMID 19029900, PMC 2680128 (freier Volltext).
- B. Crespi, P. Stead, M. Elliot: Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia. In: Proc. Natl. Acad. Sci. USA. 107 Suppl 1, Januar 2010, S. 1736–1741, doi:10.1073/pnas.0906080106, PMID 19955444, PMC 2868282 (freier Volltext).
- A. Reis, A. Rauch: Chromosomale Ursachen der geistigen Behinderung. In: Medizinische Genetik. 21 (2009), S. 237–245, doi:10.1007/s11825-009-0166-7
- R. Wimmer, H. Seidel: Chromosomale Mikrodeletionen als Ursache pädiatrischer Krankheitsbilder. In: Medizinische Genetik. 20 (2008), S. 348–356, doi:10.1007/s00112-008-1697-8
Weblinks
- Chromosome 1q21.1 Deletion Syndrome, 1.35-MB in der OMIM-Datenbank der Johns Hopkins University
- 1q21.1 recurrent microdeletion (susceptility locus for neurodevelopmental disorders) bei DECIPHER
- Brunet u. a.: BAC array CGH in patients with Velocardiofacial syndrome-like features reveals genomic aberrations on chromosome region 1q21.1; BMC Medical Genetics 2009, 10, S. 144; doi:10.1186/1471-2350-10-144
- Unique: 1q21.1 microdeletions. (PDF; 719 kB) In: www.rarechromo.org. Rare Chromosome Disorder Support Group, 2009, abgerufen am 2. Februar 2011 (englisch).
- GeneReviews NCBI Bookshelf
- Orpha.net
- Blogs über Kinder mit 1q21.1-Deletionssyndrom Chrissy, Laura and Niko
Einzelnachweise
- Unique (Memento des Originals vom 2. September 2011 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- H. C. Mefford, A. J. Sharp, C. Baker u. a.: Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. In: N Engl J Med. Band 359, Nr. 16, Oktober 2008, S. 1685–1699, doi:10.1056/NEJMoa0805384, PMID 18784092, PMC 2703742 (freier Volltext).
- H. Stefansson, D. Rujescu, S. Cichon u. a.: Large recurrent microdeletions associated with schizophrenia. In: Nature. Band 455, Nr. 7210, September 2008, S. 232–236, doi:10.1038/nature07229, PMID 18668039, PMC 2687075 (freier Volltext).
- Jennifer L. Stone, Michael C. O Donovan u. a.: Rare chromosomal deletions and duplications increase risk of schizophrenia. In: Nature. 455, 2008, S. 237–241, doi:10.1038/nature07239.
- M. Velinov, N. Dolzhanskaya: Clavicular pseudoarthrosis, anomalous coronary artery and extra crease of the fifth finger-previously unreported features in individuals with class II 1q21.1 microdeletions. In: Eur J Med Genet. Band 53, Nr. 4, 2010, S. 213–216, doi:10.1016/j.ejmg.2010.05.005, PMID 20573555.
- S. J. Diskin, C. Hou, J. T. Glessner u. a.: Copy number variation at 1q21.1 associated with neuroblastoma. In: Nature. Band 459, Nr. 7249, Juni 2009, S. 987–991, doi:10.1038/nature08035, PMID 19536264, PMC 2755253 (freier Volltext).
- eine Veröffentlichung der Klinik der Leidener Universität wird erwartet
- Lina Basel-Vanagaite u. a.: An emerging 1q21.1 deletion-associated neurodevelopmental phenotype. In: Journal of Child Neurology. 26 (1), S. 113–116, doi:10.1177/0883073810377658
- Douglas F. Levinson u. a.: Copy Number Variants in Schizophrenia: Confirmation of Five Previous Findings and New Evidence for 3q29 Microdeletions and VIPR2 Duplications. In: Am J Psychiatry. 2011; 168, S. 302–316; doi:10.1176/appi.ajp.2010.10060876.
- Masashi Ikeda u. a.: Copy Number Variation in Schizophrenia in the Japanese Population. In: Biological Psychiatry. Volume 67, Issue 3, S. 283–286 (1 February 2010) doi:10.1016/j.biopsych.2009.08.034.
- Junyan Li u. a.: Common Variants in the BCL9 Gene Conferring Risk of Schizophrenia. In: Arch Gen Psychiatry. 2011;68(3), S. 232–240. doi:10.1001/archgenpsychiatry.2011.1.