Non-Compaction-Kardiomyopathie
Als (isolierte) Non-Compaction-Kardiomyopathie (NCCM, synonym Isolated Noncompaction of Ventricular Myocardium, INVM) wird eine seltene, genetisch bedingte Störung der Herzmuskel-Verdichtung (primäre Kardiomyopathie) während der embryonalen Organentwicklung bezeichnet, die 1984 erstmals beschrieben wurde.[1] Das klinische Krankheitsbild umfasst Zeichen der Herzinsuffizienz, Herzrhythmusstörungen sowie Komplikationen durch Thrombemboliebildung. Die Diagnose erfolgt mittels Echokardiografie oder Magnetresonanztomographie. Die Behandlung ist symptomatisch, eine ursächliche Therapie ist nicht möglich.
| Klassifikation nach ICD-10 | |
|---|---|
| I42.8 | Sonstige Kardiomyopathien |
| ICD-10 online (WHO-Version 2019) | |
Entstehung
Das menschliche Herz durchläuft in der 5. bis 8. Schwangerschaftswoche einen Prozess der Gewebsverdichtung, bei dem sich Gewebsbrücken (Trabekel) und Nischen (Recessus) des Herzmuskelgewebes zurückbilden und sich der Muskel (das Myokard) kompaktiert. Als pathogenetische Ursache der Non-Compaction-Kardiomyopathie wird eine Störung in diesem komplexen Prozess postuliert. Auch eine Trennung (Dissektion) des Myokards, Einrisse oder Stoffwechselstörungen des Gewebes werden diskutiert. Der Herzmuskel ist schwammartig aufgetrieben.
Es wurden sowohl sporadisches Auftreten als auch familiäre Häufungen (bis zu 40 %) beschrieben. Es konnten verschiedene Mutationen des Gens Tafazzin (Xq28), ein Enzym des Cardiolipin-Stoffwechsels, nachgewiesen werden. Neben diesen x-chromosomal gebundenen Mutationen (siehe auch Barth-Syndrom) sind auch autosomal-dominante Vererbungen anderer Proteine (Dystrobrevin (DTAN), LIM-Domain-binding-Proteine und Sarkomer-Proteine) beschrieben.
Durch die unregelmäßige Muskelstruktur kommt es zu regionalen Unterversorgungen (Mikrozirkulationsstörungen) und Fibrosierungen des Muskels, woraus sich die Entwicklung der Herzinsuffizienz und der elektrischen Leitungsstörungen ergibt. Durch die turbulente Störung des Blutflusses (vgl. Virchow-Trias) findet eine Thromben-Bildung statt.
Häufigkeit
Das Krankheitsbild wird vermutlich in vielen Fällen nicht erkannt. Die Inzidenz in der Bevölkerung wird auf 0,05 bis 0,25 % geschätzt, ist aber letztlich unklar. Die NCCM ist bei Kindern (nach der dilatativen und hypertrophen CM) die dritthäufigste primäre Kardiomyopathie.
Klinisches Bild
Das klinische Erscheinungsbild ist von der Herzinsuffizienz (Herzschwäche) dominiert. Der Beschwerdebeginn ist sehr variabel, liegt aber meist in der Lebensmitte. Embolische Komplikationen (TIA, Schlaganfall etc.) treten gehäuft auf. Im EKG können vielfältige, meist unspezifische Rhythmusstörungen (ST-Strecken- und T-Wellenveränderungen, Schenkelblöcke, Tachykardien, Vorhofflimmern, WPW-Syndrom) auftreten.
Diagnose
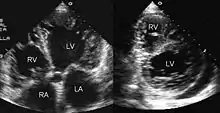
Die Diagnostik der Wahl ist die Herzultraschalluntersuchung (Echokardiografie). Es wurden diagnostische Kriterien der NCCM definiert. Eine Alternative stellt das Kardio-MRT (Magnetresonanztomographie) dar. Als cut-off gilt ein Verhältnis >2,3 zwischen nicht-kompaktem Myokard und kompakten Myokard. Die Laborparameter der Herzdiagnostik sind im Allgemeinen normal. Wegen einer möglichen familiären Häufung werden Screening-Untersuchungen der Familien empfohlen.
Behandlung und Prognose
Die Behandlung erfolgt symptomorientiert. Bei anhaltenden Tachykardien und anderen Rhythmusstörungen kann eine ICD-Implantation sinnvoll sein, die von der American Heart Association auch zur Primärprophylaxe empfohlen wird. Bei Vorhofflimmern, eingeschränkter linksventrikulärer Funktion und nachgewiesenen Thromben im Herzen ist eine gerinnungshemmende Behandlung (Antikoagulation) angezeigt. Es sind Fälle von Herztransplantationen beschrieben.
Die Datenlage zur Prognose ist unvollständig. Während sie sich bei asymptomatischen Patienten eher als günstig darstellt, ist sie beim klinischen Auftreten der NCCM ernst. Sedaghat-Hamedani et al. haben gezeigt, dass LVNC-Patienten mit Herzinsuffizienz, eine schlechtere Prognose im Vergleich zu Patienten mit dilatativer Kardiomyopathie (DCM) haben.[2]
Literatur
- R. Engberding, C. Stöllberger, P. Ong, T. Yelbuz, B. Gerecke, G. Breithardt: Isolierte Noncompaction-Kardiomyopathie Dtsch Arztebl Int 2010; 107(12), S. 206–213.
- B. C. Weiford, V. D. Subbarao, K. M. Mulhern: Noncompaction of the ventricular myocardium. In: Circulation. 2004 Jun 22;109(24), S. 2965–2971. Review. PMID 15210614
- F. Ichida: Left ventricular noncompaction. In: Circ J. 2009 Jan;73(1), S. 19–26. PMID 19057090
Einzelnachweise
- R. Engberding, F. Bender: Identification of a rare congenital anomaly of the myocardium by two-dimensional echocardiography: persistence of isolated myocardial sinusoids. In: Am J Cardiol. 1984 PMID 6731322.
- Sedaghat-Hamedani F, Haas J, Zhu F, Geier C, Kayvanpour E, Liss M, Lai A, Frese K, Pribe-Wolferts R, Amr A, Li DT, Shirvani Samani O, Carstensen A, Huang K, Zeng Q, Cheng L, Fehlmann T, Ehlermann Ph, Keller A, Dieterich C, Streckfuß-Bömeke K, Liao Y, Gotthardt M, Katus HA, Meder B: Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. In: European Heart Journal. Februar. doi:10.1093/eurheartj/ehx545. PMID 29029073.
Weblinks
- Non-Compaction-Kardiomyopathie. In: Orphanet (Datenbank für seltene Krankheiten).