Rothmund-Thomson-Syndrom
Das Rothmund-Thomson-Syndrom (RTS) ist eine autosomal-rezessiv vererbte Hautkrankheit (Genodermatose).
| Klassifikation nach ICD-10 | |
|---|---|
| Q82.8 | Sonstige näher bezeichnete angeborene Fehlbildungen der Haut |
| ICD-10 online (WHO-Version 2019) | |
Symptome und Charakterisierung
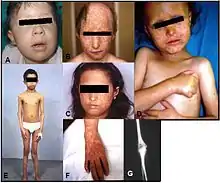
Die Erkrankung ist bei den betroffenen Patienten durch einen charakteristischen Ausschlag im Gesicht (Poikilodermie) gekennzeichnet. Die Poikilodermie ist das Leitsymptom des Rothmund-Thomson-Syndroms und dient zur Unterscheidung vom RAPADILINO-Syndrom.[1] Zur Poikilodermie kommen noch Kleinwüchsigkeit, spärliches Kopfhaar, spärliche oder fehlende Wimpern und/oder Augenbrauen, juveniler Katarakt, Fehlbildungen des Skeletts, Akromikrie, radiale Klumphand (engl. radial ray defect), vorzeitiges Altern, sowie eine Prädisposition für Krebs.[2]
Es wird zwischen zwei klinischen Unterformen des Rothmund-Thomson-Syndrom unterschieden. Typ 1 (RTS1) ist durch juvenile Katarakte, Poikilodermie und ektodermale Dysplasie gekennzeichnet, während beim Typ 2 (RTS2) Poikilodermie, kongenitale Knochendeformationen und ein erhöhtes Risiko für Osteosarkome in der Jugend und Hautkrebs im späteren Leben auftreten. Die Skelettfehlbildungen sind teilweise deutlich sichtbar ausgeprägt, wie beispielsweise prominente Stirn, Sattelnase und radiale Klumphand, teilweise aber auch nur radiologisch sichtbar.[2]
Epidemiologie
Das Rothmund-Thomson-Syndrom ist eine sehr seltene Krankheit. Über die Prävalenz liegen keine zuverlässigen Daten vor. In der medizinischen Literatur sind bis zum Jahr 2010 etwa 300 Patienten aufgeführt.[3][4][2]
Dem autosomal-rezessiven Erbgang entsprechend sind die meisten Fälle von RTS keine Einzelfälle, sondern treten familiär gehäuft, überwiegend in blutsverwandten Familien oder kleinen Gemeinden auf.[5][6][7] Die Trägerfrequenz der Mutation ist unbekannt.[2]
Von der Erkrankung können beide Geschlechter betroffen sein. Die Geschlechterverteilung von RTS ist – aufgrund der geringen Fallzahl – noch nicht gesichert. Es gibt Berichte über eine Gleichverteilung, eine weibliche Dominanz von 1,4:1, und eine männliche Dominanz von 2:1.[8][2] RTS-Fälle wurden in allen Ethnien beschrieben. Ein Gründereffekt wurde bisher in keiner Population entdeckt, auch wenn bestimmte Mutationen innerhalb einer definierten Population existieren können.[2]
Genetik
Das Rothmund-Thomson-Syndrom wird auf autosomal-rezessivem Weg vererbt und ist genetisch betrachtet heterogen. RTS2 wird durch homozygote oder durch compound-heterozygote Mutationen im RECQL4-Gen, das für eine Helikase codiert, verursacht. Missense-Mutationen sind dabei wesentlich seltener als Frameshift-, Nonsense- und Splice-Site-Mutationen. Defekte im RECQL4-Gen sind bei etwa 60 bis 65 % aller RTS-Patienten die Krankheitsursache (RTS2).[2]
Die Ätiologie von RTS1 ist dagegen bisher noch unbekannt.[2]
Diagnose
Bedingt durch die unspezifischen Symptome von RTS, gibt es keine klinischen Kriterien oder eine Bewertungstabelle (score), um die Diagnose von RTS in eindeutig, wahrscheinlich oder möglich einzuteilen. Die klinische Diagnose basiert gegenwärtig auf dem Zeitpunkt des Beginns der Erkrankung, ihrem Fortschreiten und dem Auftreten der Poikilodermie. Nach Wang und Plon kann die Diagnose wahrscheinlich RTS dann gestellt werden, wenn der Hautausschlag atypisch ist und zwei der nachfolgenden Kriterien erfüllt sind: dünne Behaarung an Kopf, Augenbrauen und Wimpern; Minderwuchs; angeborene Knochendeformationen wie z. B. Radiusaplasie, (einschließlich solcher, die nur durch bildgebende Verfahren festgestellt werden können); abnormale Zähne und Nägel; Hyperkeratose; Katarakt und Krebs.[9]
Speziell bei Patienten mit einem Osteosarkom und Hautveränderungen sollte die Möglichkeit eines Rothmund-Thomson-Syndroms als Krankheitsursache mit in Betracht gezogen werden.[10]
Molekulardiagnostisch kann bei Patienten mit RTS2 – etwa zwei Drittel aller RTS-Patienten – durch die DNA-Analyse des RECQL4-Gens die Diagnose gesichert werden. Dabei ist zu beachten, dass auch das Baller-Gerold-Syndrom und das RAPADILINO-Syndrom mit Mutationen von RECQL4 assoziiert sind und teilweise sehr ähnliche Symptome aufweisen.[11][12][13][2]
Therapie
Die Behandlung des Rothmund-Thomson-Syndroms erfolgt weitgehend symptomatisch und, wegen der Komplexität der Erkrankung, interdisziplinär zwischen Dermatologen, Augenärzten, Orthopäden, Chirurgen und Onkologen.[2] Stammzelltransplantationen wurden bisher erst an zwei Patienten durchgeführt (Stand 2010). In einem Fall eine allogene Knochenmarktransplantation,[14] in einem anderen eine mit Nabelschnurblutstammzellen.[15][2]
Prognose
Die Lebenserwartung der Patienten ist, auch wenn ein vorzeitiges Altern sichtbar ist, weitgehend normal, wenn die Krebserkrankungen rechtzeitig erkannt und behandelt werden. Im Fall von Osteosarkomen bei RTS-Patienten ist die 5-Jahres-Überlebensrate – wie bei Osteosarkom-Patienten ohne RTS auch – im Bereich von 60 bis 70 %.[16]
Erstbeschreibung

Das Rothmund-Thomson-Syndrom wurde erstmals 1868 von dem deutschen Augenarzt August von Rothmund[5] beschrieben. Der britische Arzt Matthew Sydney Thomson (1894–1969) veröffentlichte 1936 eine Abhandlung über zwei Fälle von Poikiloderma Congenitale (erbliche Poikilodermie)[6], eine Krankheit die mit der von Rothmund beschriebenen identisch war und die später die Namen der beiden Ärzte bekam.
Weiterführende Literatur
- T. Simon, J. Kohlhase, C. Wilhelm, M. Kochanek, B. De Carolis, F. Berthold: Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review. In: American journal of medical genetics. Part A Band 152A, Nummer 6, Juni 2010, S. 1575–1579, ISSN 1552-4833. doi:10.1002/ajmg.a.33427. PMID 20503338. (Review).
- Y. Liu: Rothmund-Thomson syndrome helicase, RECQ4: on the crossroad between DNA replication and repair. In: DNA repair Band 9, Nummer 3, März 2010, S. 325–330, ISSN 1568-7856. doi:10.1016/j.dnarep.2010.01.006. PMID 20096650. (Review).
- G. Stinco, G. Governatori, P. Mattighello, P. Patrone: Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: case report and published work review. In: The Journal of Dermatology Band 35, Nummer 3, März 2008, S. 154–161, ISSN 0385-2407. doi:10.1111/j.1346-8138.2008.00436.x. PMID 18346259. (Review).
- T. D. Roinioti, P. K. Stefanopoulos: Short root anomaly associated with Rothmund-Thomson syndrome. In: Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics Band 103, Nummer 1, Januar 2007, S. e19–e22, ISSN 1528-395X. doi:10.1016/j.tripleo.2006.07.021. PMID 17178481. (Review).
- R. K. Mak, W. A. Griffiths, J. E. Mellerio: An unusual patient with Rothmund-Thomson syndrome, porokeratosis and bilateral iris dysgenesis. In: Clinical and Experimental Dermatology Band 31, Nummer 3, Mai 2006, S. 401–403, ISSN 0307-6938. doi:10.1111/j.1365-2230.2006.02080.x. PMID 16681588. (Review).
- L. Larizza, I. Magnani, G. Roversi: Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. In: Cancer Letters Band 232, Nummer 1, Januar 2006, S. 107–120, ISSN 0304-3835. doi:10.1016/j.canlet.2005.07.042. PMID 16271439. (Review).
- J. M. el-Khoury, S. N. Haddad, N. G. Atallah: Osteosarcomatosis with Rothmund-Thomson syndrome. In: The British journal of radiology Band 70, Februar 1997, S. 215–218, ISSN 0007-1285. PMID 9135453. (Review).
- A. Leonard, A. W. Craft, C. Moss, A. J. Malcolm: Osteogenic sarcoma in the Rothmund-Thomson syndrome. In: Medical and pediatric oncology Band 26, Nummer 4, April 1996, S. 249–253, ISSN 0098-1532. doi:10.1002/(SICI)1096-911X(199604)26:4<249::AID-MPO5>3.0.CO;2-J. PMID 8600336. (Review).
- C. A. Drouin, E. Mongrain, D. Sasseville, H. L. Bouchard, M. Drouin: Rothmund-Thomson syndrome with osteosarcoma. In: Journal of the American Academy of Dermatology Band 28, Nummer 2 Pt 2, Februar 1993, S. 301–305, ISSN 0190-9622. PMID 8436644. (Review).
- E. M. Vennos, M. Collins, W. D. James: Rothmund-Thomson syndrome: review of the world literature. In: Journal of the American Academy of Dermatology Band 27, Nummer 5 Pt 1, November 1992, S. 750–762, ISSN 0190-9622. PMID 1430398. (Review).
- C. Moss: Rothmund-Thomson syndrome: a report of two patients and a review of the literature. In: British Journal of Dermatology Band 122, Nummer 6, Juni 1990, S. 821–829, ISSN 0007-0963. PMID 2196075. (Review).
Einzelnachweise
- Orphanet: RAPADILINO-Syndrom. Abgerufen am 9. Januar 2011
- L. Larizza, G. Roversi, L. Volpi: Rothmund-Thomson syndrome. In: Orphanet Journal of Rare Diseases Band 5, 2010, S. 2, ISSN 1750-1172. doi:10.1186/1750-1172-5-2. PMID 20113479. PMC 2826297 (freier Volltext). (Review). (Open Access)
- E. M. Vennos, M. Collins, W. D. James: Rothmund-Thomson syndrome: review of the world literature. In: Journal of the American Academy of Dermatology Band 27, Nummer 5 Pt 1, November 1992, S. 750–762, ISSN 0190-9622. PMID 1430398. (Review).
- E. M. Vennos, W. D. James: Rothmund-Thomson syndrome. In: Dermatologic Clinics Band 13, Nummer 1, Januar 1995, S. 143–150, ISSN 0733-8635. PMID 7712640. (Review).
- A. Rothmund: Ueber Cataracte in Verbindung mit einer eigenthümlichen Hautdegeneration. In: Archiv für Ophtalmologie Band 14, 1868, S. 159–182. doi:10.1007/BF02720945
- M. S. Thomson: Poikiloderma Congenitale: Two Cases for Diagnosis. In: Proceedings of the Royal Society of Medicine Band 29, Nummer 5, März 1936, S. 453–455, ISSN 0035-9157. PMID 19990626. PMC 2076117 (freier Volltext).
- S. Kitao, A. Shimamoto, M. Goto, R. W. Miller, W. A. Smithson, N. M. Lindor, Y. Furuichi: Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. In: Nature genetics Band 22, Nummer 1, Mai 1999, S. 82–84, ISSN 1061-4036. doi:10.1038/8788. PMID 10319867.
- S. Hsu: Rothmund-Thomson Syndrome. In: e-medicine vom 13. Oktober 2009
- L. L. Wang und S. E. Plon: Rothmund-Thomson Syndrome. R. A. Pagon u. a. (Herausgeber): GeneReviews Seattle, 1993–1999, PMID 20301415.
- L. A. Pujol, R. P. Erickson, R. A. Heidenreich, C. Cunniff: Variable presentation of Rothmund-Thomson syndrome. In: American journal of medical genetics Band 95, Nummer 3, November 2000, S. 204–207, ISSN 0148-7299. PMID 11102924.
- M. Debeljak, A. Zver, J. Jazbec: A patient with Baller-Gerold syndrome and midline NK/T lymphoma. In: American journal of medical genetics. Part A Band 149A, Nummer 4, Februar 2009, S. 755–759, ISSN 1552-4833. doi:10.1002/ajmg.a.32736. PMID 19291770.
- Y. Sznajer, H. A. Siitonen, G. Roversi, C. Dangoisse, M. Scaillon, F. Ziereisen, S. Tenoutasse, M. Kestilä, L. Larizza: Atypical Rothmund-Thomson syndrome in a patient with compound heterozygous mutations in RECQL4 gene and phenotypic features in RECQL4 syndromes. In: European Journal of Pediatrics Band 167, Nummer 2, Februar 2008, S. 175–181, ISSN 1432-1076. doi:10.1007/s00431-007-0447-6. PMID 17372760.
- R. Kellermayer, H. A. Siitonen, K. Hadzsiev, M. Kestilä, G. Kosztolányi: A patient with Rothmund-Thomson syndrome and all features of RAPADILINO. In: Archives of Dermatology Band 141, Nummer 5, Mai 2005, S. 617–620, ISSN 0003-987X. doi:10.1001/archderm.141.5.617. PMID 15897384.
- C. Rizzari, D. Bacchiocchi, A. Rovelli, A. Biondi, A. Cantu'-Rajnoldi, C. Uderzo, G. Masera: Myelodysplastic syndrome in a child with Rothmund-Thomson syndrome: a case report. In: Journal of pediatric hematology/oncology : official journal of the American Society of Pediatric Hematology/Oncology Band 18, Nummer 1, Februar 1996, S. 96–97, ISSN 1077-4114. PMID 8556381.
- M. A. Broom, L. L. Wang, S. K. Otta, A. P. Knutsen, E. Siegfried, J. R. Batanian, M. E. Kelly, M. Shah: Successful umbilical cord blood stem cell transplantation in a patient with Rothmund-Thomson syndrome and combined immunodeficiency. In: Clinical genetics Band 69, Nummer 4, April 2006, S. 337–343, ISSN 0009-9163. doi:10.1111/j.1399-0004.2006.00592.x. PMID 16630167.
- M. J. Hicks, J. R. Roth, C. A. Kozinetz, L. L. Wang: Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. In: Journal of clinical oncology : official journal of the American Society of Clinical Oncology Band 25, Nummer 4, Februar 2007, S. 370–375, ISSN 1527-7755. doi:10.1200/JCO.2006.08.4558. PMID 17264332.
Weblinks
- Rothmund-Thomson-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- RTS Rothmund-Thomson-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).