Pantothenatkinase-assoziierte Neurodegeneration
Die Pantothenatkinase-assoziierte Neurodegeneration (PKAN), auch als Hallervorden-Spatz-Syndrom (HSS) bezeichnet, ist eine sehr seltene neuro-degenerative Erkrankung, bei der sich insbesondere in den Basalganglien (Globus Pallidus und Substantia nigra) erhöhte Mengen an Eisen nachweisen lassen. Daher wird sie der Gruppe von Erkrankungen mit dem Namen Neurodegeneration mit Eisenablagerung im Gehirn (NBIA, englisch Neurodegeneration with Brain Iron Accumulation) untergeordnet und ist deren zweithäufigste Form.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| G23.0 | Hallervorden-Spatz-Syndrom
Pigmentdegeneration des Pallidums |
| ICD-10 online (WHO-Version 2019) | |
Ursprünglich, als nur PKAN oder das HSS bekannt war, galt NBIA als synonym für HSS. Heute gehören mindestens 10 Erkrankungen zu dieser Gruppe NBIA.[2]
Synonyme sind NBIA1 und Neurodegeneration mit Eisenspeicherung im Gehirn Typ 1.
Als autosomal-rezessiv vererbliche Erkrankung aus der Gruppe der Neuroaxonalen Dystrophien wurde PKAN erstmals 1922 von Julius Hallervorden und Hugo Spatz beschrieben.[3][4]
Vorkommen
Die Prävalenz wird auf 1–3 Betroffene je 1 Million Menschen geschätzt.[1] In Deutschland leben mindestens 45 Menschen (ohne Dunkelziffer) mit NBIA (Stand: 2008).
Ursache
Bei der Mehrzahl der erkrankten Patienten, insbesondere bei früher Erstmanifestation, lassen sich Mutationen eines für eine Pantothenatkinase (PANK2) kodierenden Gens auf dem Chromosom 20 Genort p13-p12.3 nachweisen.[5] Dieses Enzym spielt eine zentrale Rolle bei der Coenzym-A-Biosynthese. Ein Enzymdefekt führt zu einer Ansammlung von Cystein, das in Anwesenheit von Eisen (also insbesondere im Bereich der Substantia nigra und der Basalganglien) zu einem Anstieg freier Radikale führt und so eine oxidative Schädigung des Gehirns bewirkt. Es kommt zu einer exzessiven Ablagerung von Eisen und Neuromelanin.[1][6]
Einteilung
Je nach klinischem Verlauf lassen sich zwei Formen unterscheiden:
Erkrankungsverlauf
Die Erkrankung setzt gewöhnlich im Kindesalter ein, manchmal mit ausgeprägter Symptomatik schon in der ersten Lebensdekade, doch kommen auch adulte Verlaufsformen vor. Zunächst stehen extrapyramidale Bewegungsstörungen, insbesondere eine Gangstörung mit Tendenz zu häufigen Stürzen oder Beindystonie (90 %) im Vordergrund, seltener psychische Auffälligkeiten (10 %). Im Verlauf treten Bewegungsstörungen (Dystonie, Choreoathetose, Tremor) mit rigider Muskeltonuserhöhung, eine Hyperreflexie sowie eine Retardierung oder eine demenzielle Entwicklung hinzu. Dysarthrie und Dysphagie kommen ebenfalls regelmäßig im Verlauf der Erkrankung hinzu. Retinitis pigmentosa oder Optikusatrophie können daneben auftreten. Beim Erwachsenen beherrscht ein Parkinson-Plus-Syndrom mit Demenz, Hyperreflexie und prominenter Dystonie das klinische Bild. Der Krankheitsverlauf ist progressiv, das heißt, der neurologische Zustand des Patienten verschlechtert sich mit der Zeit immer weiter.
Zusatzdiagnostik
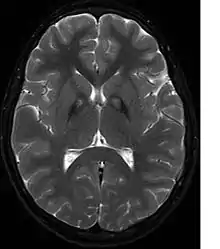
Neben dem laborchemischen Ausschluss eines Morbus Wilson, fakultativ auch einer Neuroakanthozytose, führt vor allem eine kernspintomographische Untersuchung des Gehirns zur Erhärtung der Diagnose. Hier zeigt sich in T2-gewichteten Aufnahmen im Globus pallidus charakteristischerweise eine durch Eisenablagerungen bedingte Hypointensität mit einer zentralen, vermutlich durch eine Gliose bedingten Hyperintensität. Dieser charakteristische Befund wird als „Tigerauge-Zeichen“ (englisch eye of the tiger) bezeichnet und findet sich bei allen Patienten mit PANK2-Mutationen. In einer genetischen Untersuchung lassen sich ggf. Mutationen im PANK2-Gen nachweisen. Eine sichere Diagnosestellung kann nur neuropathologisch erfolgen.
Differentialdiagnostik
Abzugrenzen sind der Morbus Wilson und andere Formen der NBIA.[1]
Therapie
Eine kausale Therapie ist bisher nicht bekannt; es gibt erste Versuche, den Enzymdefekt zu behandeln. Eisenchelatoren wie Deferoxamin sind ohne Effekt, jedoch gibt es seit 2007 erste Therapieversuche mit dem Eisenchelator Deferipron (Handelsname: Ferriprox). Auch die Tiefenhirnstimulation kommt bei Dystonie und Hyperkinesien als vorherrschender Symptomatik ggf. in Frage. Hypokinesen können mit L-DOPA behandelt werden, Hyperkinesen mit Anticholinergika, wie z. B. Trihexyphenidyl. Jedoch ist die Wirksamkeit von L-DOPA bei Patienten mit Mutationen im Gen PANK2 sehr fraglich.[9] Zur Muskelrelaxation und, damit verbunden, Schmerzlinderung werden oft Baclofen und/oder Benzodiazepine eingesetzt.
Benennung der Erkrankung
Wegen der Verstrickungen vor allem von Julius Hallervorden in das Euthanasie-Programm des Dritten Reichs ist vorgeschlagen worden,[10] die Erkrankung (basierend auf dem Gendefekt) als Pantothenatkinase-assoziierte Degeneration oder (allgemeiner gehalten) als Neurodegeneration mit Eisenablagerung im Gehirn (englisch NBIA-Syndrom – Neurodegeneration with Brain Iron Accumulation) zu bezeichnen[11]. Da weitere Erkrankungen entdeckt wurden, die wie PKAN mit einer Ablagerung von Eisen im Gehirn einhergehen, stellt NBIA heute den Oberbegriff einer Gruppe von mindestens 10 Erkrankungen dar.[12] Die Begriffe NBIA und PKAN haben sich weltweit immer mehr durchgesetzt.[13]
Literatur
- Marshall, R.D., Collins, A., Escolar, M.L. et al.: Diagnostic and clinical experience of patients with pantothenate kinase-associated neurodegeneration. In: Orphanet J Rare Dis, Bd. 14, S. 174, 2019. doi:10.1186/s13023-019-1142-1
Einzelnachweise
- Pantothenat-Kinase-assoziierte Neurodegeneration. In: Orphanet (Datenbank für seltene Krankheiten).
- Allison Gregory, Susan Hayflick: Neurodegeneration with Brain Iron Accumulation Disorders Overview. In: GeneReviews®. University of Washington, Seattle, Seattle (WA) 1993, PMID 23447832 (nih.gov [abgerufen am 9. Dezember 2021]).
- J. Hallervorden, H. Spatz: Eigenartige Erkrankung im extrapyramidalen System mit besonderer Beteiligung des Globus pallidus und der Substantia nigra: Ein Beitrag zu den Beziehungen zwischen diesen beiden Zentren. In: Zeitschrift für die gesamte Neurologie und Psychiatrie. Band 79, Nr. 1, Dezember 1922, ISSN 0303-4194, S. 254–302, doi:10.1007/BF02878455 (springer.com [abgerufen am 12. Februar 2022]).
- J. Hallervorden: Über ein familiäre Erkrankung im extrapyramidalen System. In: Deutsche Zeitschrift für Nervenheilkunde. Band 81, Nr. 1-4, Januar 1924, ISSN 0340-5354, S. 204–210, doi:10.1007/BF01669546 (springer.com [abgerufen am 12. Februar 2022]).
- Zhou u. a.: A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. in: Nature genetics, 28.2001,4 (Aug), S. 345–349. PMID 11479594 ISSN 1061-4036
- NBIA1. In: Online Mendelian Inheritance in Man. (englisch)
- Pantothenat-Kinase-assoziierte Neurodegeneration, klassische Form. In: Orphanet (Datenbank für seltene Krankheiten).
- Pantothenat-Kinase-assoziierte Neurodegeneration, atypische Form. In: Orphanet (Datenbank für seltene Krankheiten).
- Gregory, Hayflick: PKAN-Artikel bei genereviews, revidierte Version Jan. 2008
- Krankheitsbezeichnungen von NS-Ärzten
- M. Shevell: Hallervorden and history. In: The New England journal of medicine, 2.2003,1 (Jan), 348, S. 3–4. PMID 12510036 ISSN 0028-4793
- Allison Gregory, Susan Hayflick: Neurodegeneration with Brain Iron Accumulation Disorders Overview. In: GeneReviews®. University of Washington, Seattle, Seattle (WA) 1993, PMID 23447832 (nih.gov [abgerufen am 9. Dezember 2021]).
- Lawrence A. Zeidman, Dilip K. Pandey: Declining use of the Hallervorden-Spatz disease eponym in the last two decades. In: Journal of Child Neurology. Band 27, Nr. 10, Oktober 2012, ISSN 1708-8283, S. 1310–1315, doi:10.1177/0883073812449907, PMID 22832768 (nih.gov [abgerufen am 9. Dezember 2021]).