Osteopoikilose
Die Osteopoikilose (Osteopathia condensans disseminata, Osteopoikilie) ist eine seltene, gutartige, meist zufällig entdeckte Knochenfehlbildung. Dabei finden sich im Becken und in den Meta- und Epiphysen sowie in den Hand- und Fußwurzelknochen Cluster-artig viele, unregelmäßige, runde bis ovale Verdichtungszonen der Spongiosa von wenigen Millimetern bis Zentimetern. Die Veränderungen finden sich seltener in der Diaphyse. Oft ist das Verteilungsmuster symmetrisch. Schädel und Wirbelsäule sind selten betroffen. Die Herde gehen ohne klare Grenze in die Umgebung über. Die Kompakta ist nicht betroffen.
| Klassifikation nach ICD-10 | |
|---|---|
| Q78.8 | Sonstige näher bezeichnete Osteochondrodysplasien
Osteopoikilie |
| ICD-10 online (WHO-Version 2019) | |
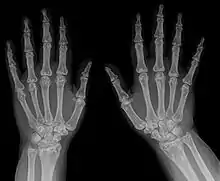
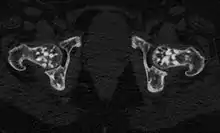
Histologisch liegt den Knochenveränderungen lamellärer Knochen zugrunde, der dem der Kompakta entspricht. Es wird vermutet, dass es sich um alte Remodellierungen handelt, die vor Umwandlung des lamellären Knochens in spongiösen Knochen inaktiviert wurden. Histologisch sind die Verdichtungen nicht von solitären Kompaktainseln zu unterscheiden.[1] Störungen des Mineralstoffwechsels sind nicht mit dem Krankheitsbild verbunden. Die Herde sind in der Skelettszintigraphie meist nicht zu sehen, was darauf hinweist, dass kein vermehrter Knochenumbau stattfindet.
Nach der Erstbeschreibung durch den Radiologen und Chirurgen Heinrich Albers-Schönberg[2] 1915 wurden bis 2005 geschätzt nur max. 400 Fälle veröffentlicht. Eine amerikanische Studie schätzt die Inzidenz auf 0,1 Fall pro Million.[3] Eine sichere Aussage über die Häufigkeit ist allerdings nicht möglich, weil die Osteopoikilose meist symptomlos verläuft. Laut dieser Studie sind Männer etwas häufiger betroffen als Frauen. In mehreren Fällen wurde eine familiäre Häufung beobachtet, wobei ein autosomal dominanter Erbgang vorzuliegen scheint, während andere Fälle sporadisch auftreten.
In einer der größten Untersuchungen zur Osteopoikilose, einer türkischen Studie von 1992,[3] fanden sich ausgehend von vier Patienten, bei denen die Osteopoikilose zufällig gefunden wurde, bei Untersuchung der Familienangehörigen schließlich 49 weitere Fälle (insgesamt 33 Männer und 20 Frauen, Verhältnis 1,65:1). Anhand der Stammbäume wurde in diesen Familien aus Ost-Anatolien und der Schwarzmeergegend eine autosomal-dominante Vererbung aufgezeigt. Bei 51 von 53 Patienten war die Osteopoikilose beidseitig. Betroffen waren vorwiegend die Metaphysen, sowie die benachbarten Knochenregionen, ohne Unterschied zwischen distalen und proximalen Metaphysen. Die Skleroseherde fanden sich in absteigender Häufigkeit in den Fingerknochen (Phalangen, 100 %), Handwurzelknochen (Carpalia, 97,4 %), Mittelhandknochen (Metacarpalia, 92,3 %), Zehenknochen (Phalangen, 87,2 %), Mittelfußknochen (Metatarsalia, 84,4 %), Fußwurzelknochen (Tarsalia, 84,6 %), gefolgt von Becken (74,4 %), Oberschenkelknochen (Femur, 74,4 %), Speiche (Radius, 66,7 %), Elle (Ulna, 66,7 %), Kreuzbein (Sacrum, 58,9 %), Oberarmknochen (Humerus, 28,2 %), Schienbein (Tibia 20,5 %) und Wadenbein (Fibula, 12,8 %).
In dieser Studie fanden sich zwischen einem und tausend Skleroseherde pro Knochen, wobei die Anzahl mit dem Alter zunahm, ebenso die Dichte (Sklerosierung). In den Beckenknochen fand sich die höchste Anzahl von Skleroseherden. Die Läsionen maßen zwischen 1 × 1 mm und 12 × 16 mm. Sie wurden als linear, elliptisch oder rund beschrieben, und waren oft entlang gedachter längsverlaufender Linien im Knochen angeordnet.
Eine ätiologische Verwandtschaft zur Osteopathia striata[4] und zur Melorheostose[5] wird diskutiert, da die Krankheitsbilder auch gemeinsam bei einem Menschen auftreten können. Neben den genannten Osteodysplasien sind weitere Begleiterkrankungen der Osteopoikilose beschrieben, darunter die Buschke-Ollendorff-Syndrom (Dermatofibrosis lenticularis disseminata)[6]), die etwa bei 10 % der Patienten mit Osteopoikilose anzutreffen ist und neben den Knochenveränderungen multiple kleine weißlich-gelbliche kutane und subkutane Knötchen aufweist. Daneben werden auch die Dakryozystitis (Günal-Seber-Başaran-Syndrom[7]) und Multiple kartilaginäre Exostosen bei Osteopoikilose beobachtet.
Die Knochenveränderungen können schon im Kleinkindalter beobachtet werden. Sie können im Laufe der Zeit zunehmen, kleiner werden, oder auch ganz verschwinden. Pathologische Frakturen treten nicht auf, die Osteopoikilose ist asymptomatisch. Eine Behandlung ist daher nicht notwendig, und auch nicht möglich. Extrem selten wurde eine maligne Entartung beschrieben.[1]
Quellen
- G. Haberhauer, M. Skoumal: Osteopoikilose. Journal für Mineralstoffwechsel 2005
- K. Bohndorf, H.Imhof: Radiologische Diagnostik der Knochen und Gelenke Thieme Stuttgart, New York 1998 ISBN 3-13-110982-3
- Heuck: Radiologie der Knochen- und Gelenkerkrankungen. Thieme Verlag, 1997, ISBN 3-13-107071-4
Einzelnachweise
- J. A. Herring: Tachdijan's Pediatric Orthopaedics. (3. Auflage, in drei Bänden) W. B. Saunders Company, Philadelphia (USA) 2002
- H. Albers-Schönberg: Eine seltene, bisher nicht bekannte Strukturanomalie des Skelettes. Fortschritte auf dem Gebiete der Röntgenstrahlen und der Nuklearmedizin 1915/1916; 23: 174–5.
- I. T. Benli, S. Akalin, E. Boysan et al.: Epidemiological, clinical and radiological aspects of osteopoikilosis. J Bone Joint Surg 1992; 74-Br: 504
- S. Ghai, R. Sharma, S. Ghai: Mixed sclerosing bone dysplasia--a case report with literature review. Clin Imaging. 2003 May-Jun;27(3):203-5.
- R. Happle: Melorheostosis may originate as a type 2 segmental manifestation of osteopoikilosis. Am J Med Genet A. 2004 Mar 15;125A(3):221-3.
- A. Buschke, H. Ollendorf: Ein Fall von Dermatofibrosis lenticularis disseminata und Osteopathia condensans disseminata. Dermatologische Wochenschrift 1928; 86: 257–62
- I. Gunal, S. Seber, N. Basaran, S. Artan, K. Gunal, E. Gokturk: Dacryocystitis associated with osteopoikilosis. Clin. Genet. 44: 211-213, 1993. PubMed ID: 8261652