Multiple kartilaginäre Exostosen
Multiple kartilaginäre Exostosen ist die Bezeichnung für eine autosomal-dominant vererbte Erkrankung mit zahlreichen gutartigen knorpelbedeckten Knochentumoren. Da es sich um Osteochondrome handelt, die mit hyalinem Knorpel statt Faserknorpel bedeckt sind, ist die Bezeichnung „kartilaginäre“ Exostose eigentlich nicht korrekt.[1] Mit einer Inzidenz von etwa 1:50.000 zählt die Exostosenkrankheit zu den häufigsten Knochentumorerkrankungen,[2] Frauen und Männer sind gleich häufig betroffen. Bei etwa 70 % der Patienten liegt eine familiäre Form vor, etwa 30 % haben eine sporadische Form, also eine Neumutation.
| Klassifikation nach ICD-10 | |
|---|---|
| Q78.6 | Angeborene multiple Exostosen - Multiple kartilaginäre Exostosen |
| ICD-10 online (WHO-Version 2019) | |
Zum Zeitpunkt der Geburt sind Exostosen noch sehr selten, sie entstehen vor allem in der Kindheit und Jugend, bis zum Schluss der Wachstumsfugen. In dieser Zeit nehmen die Exostosen auch an Größe zu. Nach Ende des Wachstums bilden sich keine weiteren Exostosen, und eine spätere Größenzunahme ist dann ein Zeichen einer malignen Transformation (Entartung), d. h. Umwandlung in einen bösartigen Knochentumor. In etwa 3 – 5 % tritt ein sekundäres Chondrosarkom auf.
Die Exostosenkrankheit kann selten völlig asymptomatisch sein, die Ausprägung (Phänotyp) ist aber hochgradig variabel, selbst in einer Familie. Bei etwa 19 % finden sich maximal fünf Exostosen, bei etwa 19 % sind es mehr als zwanzig.
Ganz generell werden die klinischen Probleme durch Deformitäten und funktionelle Einschränkungen geprägt, woraus die klinische Einteilung der Ausprägung nach Pedrini in drei Klassen und jeweils zwei Untergruppen resultiert:[3]
- 1 – Keine Deformitäten und keine funktionellen Einschränkungen (38 %, 1A – maximal fünf Exostosen, 1B- mehr als fünf Exostosen)
- 2 – Deformitäten, aber keine funktionellen Einschränkungen (37 %, 2A – maximal fünf Deformitäten, 2B – mehr als fünf Deformitäten)
- 3 – Deformitäten und funktionelle Einschränkungen (25 %, 3A – maximal eine funktionelle Einschränkung, 3B – mehr als eine funktionelle Einschränkung)
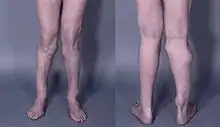
Da die Exostosen metaphysär in Nähe der Wachstumsfugen ausgehen, finden sie sich oft in Gelenknähe und führen dort zu Bewegungseinschränkungen, achsabweichendes Wachstum mit Deformierungen und Hypoplasie (Minderwuchs) einzelner Knochen bis zum generellen Kleinwuchs. Typische Fehlstellungen sind:
- Längendifferenzen im Bereich der Extremitäten (Beinlängendifferenz)
- Achsenabweichungen im Bereich der Knie und Sprunggelenke Genu valgum
- Asymmetrien des Schulter- und Beckengürtels,
- Subluxation des Hüftgelenkes infolge Herausdrängens des Hüftkopfes durch den nach medial sich exostotisch verbreiternden Schenkelhals
- Ulnarabweichung des Handgelenks aufgrund einer Ulnarverkürzung mit konsekutiver Verkrümmung der Speiche in Richtung der zu kurzen Elle mit Subluxation des Radiokarpalgelenks.[4]
Die Exostosen können aber auch in der direkten Nachbarschaft stören und Nerven, Gefäße oder Sehnen komprimieren und so Schmerzen und motorische Probleme auslösen.
Mitunter finden sich auch Foramina parietalia permagna.[5]
Genetik
Der Exostosenkrankheit liegt fast immer eine Mutation eines der beiden Gene EXT1 (65 %) oder EXT2 (25 %) zugrunde. In der Regel führen die Mutationen zu verkürzten und komplett oder teilweise funktionslosen EXT-Proteinen. Die beiden EXT-Proteine sind ubiquitär vorkommende Transmembran-Glycoproteine, die an der Verlängerung von Heparan-Sulfat-Glykosaminoglykan-Ketten (HS-GAG) der Matrix-Proteoglykanen beteiligt sind, die wiederum im Rahmen des Zellwachstums und der Differenzierung von Knorpelzellen eine wichtige Rolle spielen.
Behandlung
Die Behandlung richtet sich nach den Störungen, und umfasst Schmerztherapie, Physiotherapie und chirurgische Eingriffe zum Abtragen störender Exostosen und zur Deformitätenkorrektur. Jedoch sind die Ergebnisse nicht immer zufriedenstellend, insbesondere bei multiplen Operationen.
Synonyme
.jpg.webp)
- Ekchondrosis ossificans
- multiple Osteochondromatose
- multiple hereditäre Exostosen (MHE)
- hereditäre multiple Exostosen (HME)
- Exostosenkrankheit
- multiple Osteochondrome
- exostotische Dysplasie
- Osteoplasia exostotica
- multiple Osteomatose
- dominant erbliche chondrale Osteome
- multiple cartilaginous exostoses (engl.)
- Bessel-Hagen-Krankheit[6]
Einzelnachweise
- F. Hefti: Kinderorthopädie in der Praxis. Springer-Verlag, Berlin 1997, ISBN 3-540-61480-X.
- G. A. Schmale, E. U. Conrad, W. H. Raskind: The natural history of hereditary multiple exostoses. In: The Journal of Bone & Joint Surgery. Volume 76, Issue 7, 1994, PMID 8027127
- E. Pedrini u. a.: Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: identification of „protective“ and „risk“ factors. In: J Bone Joint Surg. 2011; 93-Am, S. 2294–2302.
- J. R. Stieber, J. P. Dormans: Manifestations of hereditary multiple exostoses. In: Journal of the American Academy of Orthopaedic Surgeons. 2005; 13 (2), S. 110–120. PMID 15850368
- Exostoses, Multiple, Type II. In: Online Mendelian Inheritance in Man. (englisch)
- Osteochondrome, multiple. In: Orphanet (Datenbank für seltene Krankheiten).