Neighbor-Joining-Algorithmus
Der Neighbor-Joining-Algorithmus ist ein mathematisches Verfahren, um Datensätze zu vergleichen und hierarchisch bifurcal (zweigabelig) anzuordnen. Dieses Verfahren wurde 1987 von Saitou und Nei vorgestellt[1] und 1988 von Studier und Keppler weiterentwickelt und vereinfacht.
Anwendung
In der Bioinformatik bezeichnet das Neighbor-Joining-Verfahren eine phänetische bottom-up Clustermethode, welche zur Erstellung von phylogenetischen Baumstrukturen verwendet wird. Hiermit soll anhand von variierenden Merkmalen in der Datenmatrix die Wahrscheinlichkeit einer Abstammungs- oder Verwandtschaftsbeziehung in einer stammbaumartigen Darstellung berechnet werden.
Normalerweise werden damit Bäume aus DNA- oder Proteinsequenzdaten oder klassisch morphologischen Datensätzen erstellt. Der Algorithmus benötigt Wissen über die Distanz zwischen zwei Paaren von Taxa (also beispielsweise Arten oder Sequenzen) in einem Baum.
Algorithmus
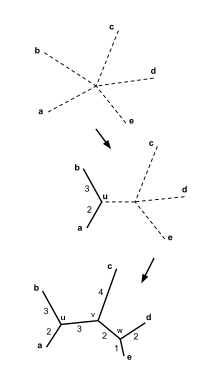
Neighbor-joining basiert meist auf dem „Minimum Evolution“-Kriterium für phylogenetische Bäume: Ausgehend von einem zunächst sternförmigen „Baum“, in dem alle Taxa mit einem „Zentrum“ verbunden sind, werden paarweise die DNA- oder Proteinsequenzen mit der geringsten genetischen Distanz ausgewählt und zu einem Ast des Baumes vereinigt. Die genetischen Distanzen der Sequenzen werden neu berechnet und wieder die nächstverwandten zu einem Ast mit zwei Taxa zusammengefügt. Dies erfolgt solange, bis alle Taxa in dem Baum eingefügt wurden und die Sternstruktur des Baumes völlig aufgelöst wurde. Im Unterschied zum UPGMA berücksichtigt Neighbor-Joining, dass die Evolutionsgeschwindigkeit nicht konstant ist: Wenn ein Taxon von allen anderen Taxa weit entfernt ist, so ist dies nicht auf einen entfernten Verwandtschaftsgrad, sondern auf beschleunigte Evolution zurückzuführen.
Der Algorithmus ist iterativ und ersetzt in jedem Schritt ein Paar der Operational Taxonomic Units (OTU) durch eine neue Sequenz. Er iteriert auf den jeweils verbleibenden Sequenzen weiter, bis es für drei verbleibende OTUs nur noch eine mögliche Topologie gibt. Danach wird die Baumstruktur erstellt.[2]
Beispiel
Folgend ist eine typische Tabelle von Distanzen zwischen Taxa angegeben, wobei die Werte rein hypothetisch, aber realistisch sind:[3] Studier und Keppler führten einen alternativen Parameter im Algorithmus ein, der als Mij bezeichnet wird. Saitou und Nei verwendeten ursprünglich zur Bestimmung der Nachbarn die “minimal sum of branches” (Sij) also die minimale Anzahl an Verzweigungen. Das Beispiel basiert auf dem Algorithmus von Studier und Keppler, welche gegen dem ursprünglichen Parameter eine Komplexitätsklasse bietet.[4]

| Mensch | Maus | Rose | Tulpe | |
|---|---|---|---|---|
| Mensch | 0 | 3 | 14 | 12 |
| Maus | 3 | 0 | 13 | 11 |
| Rose | 14 | 13 | 0 | 4 |
| Tulpe | 12 | 11 | 4 | 0 |
Da die Tabelle dreiecks-symmetrisch ist, muss die untere Hälfte nicht unbedingt gespeichert werden. Die Werte in dieser Tabelle werden als benannt.

Schritt 1: Es müssen die Durchschnittlichen Distanzen von jedem Taxon zu jedem anderen berechnet werden. Dies geschieht mit folgender Formel für die Netto-Divergenz ri:[2]

Wobei N die Anzahl der Taxa ist.
| Mensch | |
|---|---|
| Maus | |
| Rose | |
| Tulpe |




Interpretation: Die Rose besitzt in unserem Beispiel die größte Netto-Divergenz, hat also im Vergleich mit den anderen Taxa eine größere Evolutionsgeschwindigkeit durchlebt.
Schritt 2: Wir berechnen eine Zwischenmatrix M.

Wie z. B. zwischen Mensch und Maus:

| Mensch | Maus | Rose | Tulpe | |
|---|---|---|---|---|
| Mensch | −25 | −16 | −16 | |
| Maus | −25 | −16 | −16 | |
| Rose | −16 | −16 | −25 | |
| Tulpe | −16 | −16 | −25 |

Schritt 3: In dieser neu berechneten Distanzmatrix M wird nun der kleinste Wert, also die kleinste Distanz zwischen zwei Taxa, gesucht, und die gefundenen zwei Taxa werden zu einem neuen Teilbaum u = (i,j) zusammengefügt. In diesem Beispiel ergeben sich also die zwei Möglichkeiten Mensch und Maus, oder Rose und Tulpe zu einem Teilbaum zusammenzufügen. Wir entscheiden uns zunächst für Mensch und Maus.
Die Kantenlänge des Knotens zu der Verzweigung berechnet sich wie folgt:


Also Mensch zu MeMa ist gleich

Schritt 4: In der ursprünglichen Distanzmatrix wird der neue Eintrag u = MeMa angefügt:
| Mensch | Maus | Rose | Tulpe | MeMa | |
|---|---|---|---|---|---|
| Mensch | 0 | 3 | 14 | 12 | ? |
| Maus | 3 | 0 | 13 | 11 | ? |
| Rose | 14 | 13 | 0 | 4 | ? |
| Tulpe | 12 | 11 | 4 | 0 | ? |
| MeMa | ? | ? | ? | ? | 0 |
Um die Distanzen des neuen Eintrages u = (i,j) = (1,2) = (Mensch, Maus) = MeMa zu den restlichen Taxa zu berechnen, wird folgende Formel verwendet:

Wobei die Einträge i und j zu einem neuen Eintrag u zusammengefügt wird, und die Distanz zum Eintrag k ausgerechnet wird. Die Distanz zwischen Rose und dem neuen Teilbaum ist also:

Die „alten“, zusammengefügten Einträge, werden aus den Distanzmatrixen gelöscht.
| Rose | Tulpe | MeMa | |
|---|---|---|---|
| Rose | 0 | 4 | 12 |
| Tulpe | 4 | 0 | 10 |
| MeMa | 12 | 10 | 0 |
Danach werden wieder und berechnet, neu zusammengefügt und wieder von vorne angefangen. Dies wird solange wiederholt, bis nur noch zwei Taxa übrig bleiben, die dann schlussendlich verbunden werden.


Das Ergebnis unseres Beispiels lässt sich wie folgt darstellen:
| additiver Baum | Ausgabe des Phylip Programms |
|---|---|
Mensch Rose
\ /
\ 2 3 /
\ 9 /
-----------------
/ \
/ 1 1 \
/ \
Maus Tulpe
|
+----Maus ! ! +-------------Rose 1-----------------------------------------2 ! +----Tulpe ! +--------Mensch |
Einordnung
Neighbor-Joining gehört zu den expliziten Methoden. Dies bedeutet, dass bei der Berechnung der genetischen Distanzen unterschiedliche Evolutionsmodelle, also unterschiedliche Wahrscheinlichkeiten für Punktmutationen angenommen werden können. Die Richtigkeit dieser Stammbäume beruht auf der Annahme, dass die Veränderung der betrachteten Merkmale keine unbekannten Zwischenschritte enthält. Es wird also vereinfacht angenommen, dass „die Evolution keine Umwege geht“ (“minimum evolution”).
Der Neighbor-Joining-Algorithmus berechnet den Stammbaum schrittweise und findet deshalb nicht zwangsläufig die optimale Baum-Topologie mit der geringsten Verzweigungslänge. Dies beruht auf seinem Konstruktionsprinzip, als Greedy-Algorithmus.[5] Im Gegensatz zu anderen Algorithmen berechnet dieser nicht alle möglichen Bäume und wählt zum Schluss die optimalen aus, sondern verwirft schon während des Verfahrens einige Rechenwege. Obwohl der Algorithmus suboptimal ist, wurde er ausführlich getestet und findet normalerweise einen Baum, der dem Optimum relativ nahekommt.
Vorteile
Der größte Vorteil dieses Verfahrens ist seine Geschwindigkeit. Man kann es auf gewaltige Datenmengen anwenden, selbst dort, wo andere Methoden der phylogenetischen Analyse wie maximum parsimony und Maximum-Likelihood nicht mehr durchführbar sind. Im Gegensatz zum UPGMA-Algorithmus (Unweighted Pair Group Method with Arithmetic mean) zur phylogenetischen Baumrekonstruktion nimmt Neighbor-Joining nicht an, dass die Entwicklung der Abstammungslinien mit derselben Rate (siehe auch Molekulare Uhr) stattfindet und erzeugt daher infolgedessen einen unbalancierten Baum.
Literatur
- N. Saitou, M. Nei: The neighbor-joining method. A new method for reconstructing phylogenetic trees. In: Molecular Biology and Evolution. Band 4, Nr. 4, 1. Juli 1987, S. 406–425 (oxfordjournals.org).
- J. A. Studier, K. J. Keppler: A note on the neighbor-joining algorithm of Saitou and Nei. In: Molecular Biology and Evolution. Band 5, Nr. 6, 1. November 1988, S. 729–731, PMID 3221794 (mbe.oxfordjournals.org [PDF]).
- Volker Knoop, Kai Müller: Gene und Stammbäume. Ein Handbuch zur molekularen Phylogenetik. 1. Auflage. Elsevier, Spektrum Akademischer Verlag, München / Heidelberg 2006, ISBN 3-8274-1642-6.
- Olivier Gascuel, Mike Steel: Neighbor-Joining Revealed. In: Molecular Biology and Evolution. Band 23, Nr. 11, 1. November 2006, S. 1997–2000, doi:10.1093/molbev/msl072, PMID 16877499.
Quellen
- 2.2.2 Neighbour-Joining-Methode (PDF, S. 9) auf spline.de
- 3.1 neighbor-joining-Algorithmus (PDF, S. 7).
- The Neighbor-Joining Method auf icp.ucl.ac.be
- Neighbour Joining Method (Saitou and Nei, 1987) Summary (Memento des Originals vom 16. September 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. auf stat.berkeley.edu
- Kord Eickmeyer, Peter Huggins, Lior Pachter, Ruriko Yoshida: On the optimality of the neighbor-joining algorithm. In: Algorithms for Molecular Biology (AMB). Band 3, 30. April 2008, ISSN 1748-7188, S. 5, doi:10.1186/1748-7188-3-5.
Weblinks
- Quicktree – Eine Implementierung von Neighbour-Joining (Memento vom 11. März 2010 im Internet Archive)
- Das PHYLIP-Paket