Kindler-Syndrom
Das Kindler-Syndrom ist eine seltene autosomal rezessiv vererbte Hautkrankheit (Dermatose), die durch eine Mutation des KIND1-Gens verursacht wird. Die Krankheit wird in der Literatur auch als kongenitale bullöse Poikilodermie beschrieben.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| L13.8 | Sonstige näher bezeichnete bullöse Dermatosen |
| ICD-10 online (WHO-Version 2019) | |
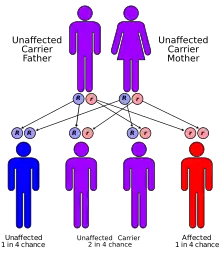
Die Krankheit gehört, wie die Porphyria cutanea tarda, die erythropoetische Protoporphyrie und die aktinische Prurigo, zu der Gruppe der Photodermatosen.[3]
Synonyme sind: Akrokeratotisches Poikiloderma; KS; Poikilodermie Typ Kindler; englisch POIKILODERMA, HEREDITARY ACROKERATOTIC;BULLOUS ACROKERATOTIC POIKILODERMA OF KINDLER AND WEARY;POIKILODERMA, CONGENITAL, WITH BULLAE, WEARY TYPE
Die Bezeichnung bezieht sich auf den Erstautor der Erstbeschreibung aus dem Jahre 1954 durch die Ärztin Theresa Kindler.[4]
Von P. Weary stammt eine weitere Beschreibung aus dem Jahre 1971.[5][6]
Diagnose
Bei Neugeborenen ist eine Blasenbildung auf der Haut zu beobachten. Die oft hypo- oder hyperpigmentierte Haut ist trocken und leicht schuppig. Dazu kommen Lichtempfindlichkeit (Photosensitivität), Atrophie und Fragilität der Haut[7] sowie Verwachsungen der Schleimhäute und Übergangsschleimhäute.[1] Bereits in der Jugend zeigt sich bei den Patienten eine Parodontose. Im Anal- und Genitalbereich der Patienten sind Schleimhauterosionen und Fissuren zu finden. Des Weiteren Urethrastenose und Phimose.[8] Im Elektronenmikroskop sind Unterbrechungen und Verzweigungen der Basalmembran sichtbar.[8]
Inzidenz
Das Kindler-Syndrom ist eine sehr seltene Erkrankung. Weltweit wurden, seit der ersten Beschreibung von Theresa Kindler, über 100 Fälle der Krankheit berichtet. Ein Cluster von 26 Patienten mit diesem Syndrom wurde bei einem Volksstamm in der Provinz Bocas del Toro im Nordwesten der Karibikküste von Panama gefunden.
Genetik
Die Ursache für das Kindler-Syndrom ist eine Mutation auf Chromosom 20, Genlocus 20p13.[9] Auf dieser Position liegt das C20orf42-Gen (KIND1), welches das Kindlin-1-Protein, auch Kindlerin oder Kindlin-Syndrom-Protein genannt, kodiert. Kindlin 1 ist ein intrazelluläres Zytoskelett-Linkerprotein, das aus 677 Aminosäuren aufgebaut ist.[8][10]
Die Abwesenheit des Kindlin-1-Proteins in der Haut führt zu mehreren Veränderungen der basalen Keratinozyten: die Zellpolarität ist aufgehoben, die Proliferation ist erheblich reduziert und viele Zellen unterliegen der Apoptose (programmierter Zelltod).[11]
Kindlin-1 ist ein Homolog des Proteins UNC-112 des Fadenwurms Caenorhabditis elegans. Dies ist ein membran-assoziiertes Struktur- / Signalprotein, das eine wichtige Rolle beim Verbinden des Aktins des Zytoskelettes mit der Extrazellulären Matrix spielt.[12]
Prognose
Die Patienten haben eine normale Lebenserwartung. Mit zunehmendem Alter verringert sich die Blasenbildung der Haut. Das Risiko an Hautkrebs oder einem Karzinom der Schleimhäute (Plattenepithelkarzinom) zu erkranken, ist allerdings signifikant höher.[13]
Literatur
- A. Hovnanian u. a.: Poikiloderma of Theresa Kindler: report of a case with ultrastructural study, and review of the literature. In: Pediatric Dermatology. 6/1989, S. 82–90. PMID 2664739
- R. C. Sharma u. a.: Kindler syndrome. In: International Journal of Dermatology. 42/2003, S. 727–732.
- E. Sadler u. a.: Novel KIND1 gene mutation in Kindler syndrome with severe gastrointestinal tract involvement. In: Archives of Dermatology. 142/2006, S. 1619–1624. PMID 17178989
- C. Has u. a.: Molecular basis of Kindler syndrome in Italy: novel and recurrent Alu/Alu recombination, splice site, nonsense, and frameshift mutations in the KIND1 gene. In: Journal of Investigative Dermatology. 126/2006, S. 1776–1783. PMID 16675959
- S. Kloeker u. a.: The Kindler syndrome protein is regulated by transforming growth factor-beta and involved in integrin-mediated adhesion. In: Journal of Biological Chemistry. 279/2004, S. 6824–6833. PMID 14634021
- L. Lennartz, C. Has, P. Lehmann: Congenital bullous poikiloderma (Kindler syndrome) – new mutation. In: Journal der Deutschen Dermatologischen Gesellschaft Bd. 10, Nr. 12, Dezember 2012, S. 919–920, doi:10.1111/j.1610-0387.2012.08050.x, PMID 23078512.
- C. Has: Kindler-Syndrom. Eine neue bullöse Dermatose. In: Der Hautarzt; Zeitschrift für Dermatologie, Venerologie, und verwandte Gebiete. Bd. 60, Nr. 8, August 2009, S. 622–626, doi:10.1007/s00105-008-1676-y, PMID 19533072 (Review).
Einzelnachweise
- B. Binder u. a.: Kongenitale bullöse Poikilodermie (Kindler-Syndrom) In: Der Hautarzt. 53/2002, S. 546–549.
- Kindler-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- P. Poblete-Gutiérrez u. a.: Hereditary photodermatoses. In: Der Hautarzt. 57/2006, S. 1067–1082.
- T. KINDLER: Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. In: The British journal of dermatology. Bd. 66, Nr. 3, März 1954, S. 104–111, PMID 13149722.
- P. E. Weary, W. F. Manley, G. F. Graham: Hereditary acrokeratotic poikiloderma. In: Archives of dermatology. Bd. 103, Nr. 4, April 1971, S. 409–422, PMID 4253719.
- Poikilodermie, akrokeratotische kongenitale, Typ Weary. In: Orphanet (Datenbank für seltene Krankheiten).
- genecards.org chromosome 20 open reading frame 42, abgerufen am 22. März 2008.
- Netzwerk Epidermolysis bullosa: Kindler-Syndrom (Memento des Originals vom 5. Januar 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis., abgerufen am 22. März 2008.
- genecards.org Chromosome 20
- Kindler syndrome. In: Online Mendelian Inheritance in Man. (englisch)
- C. Herz u. a.: Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. In: J Biol Chem. 281/2006, S. 36082–36090. PMID 17012746.
- D. H. Siegel u. a.: Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. In: Am J Hum Genet. 174/2003, S. 174–187. PMID 12789646
- G. H. Ashton u. a.: Recurrent mutations in kindlin-1, a novel keratinocyte focal contact protein, in the autosomal recessive skin fragility and photosensitivity disorder, Kindler syndrome. In: Journal of Investigative Dermatology. 2004, Band 122, S. 78–83. PMID 14962093.