Hypotrichose mit juveniler Makuladystrophie
Hypotrichose mit juveniler Makuladystrophie (HJMD oder CDH3) ist eine äußerst seltene angeborene Erkrankung, gekennzeichnet durch spärlichen Haarwuchs (Hypotrichose) von Geburt an und fortschreitende Makuladystrophie.
Häufigkeit
Ihre Gesamthäufigkeit wird auf weit weniger als 1:1.000.000 geschätzt, bislang sind nur etwa 50 bis 100 Fälle beschrieben.[1]
Ursache
Die Erkrankung wird autosomal-rezessiv vererbt. Ursächlich liegen Mutationen im CDH3 Gen vor, das für das Cadherin-3 Protein (P-Cadherin) kodiert, ein Calcium bindendes Protein, das in unterschiedlichen Geweben für den Zellkontakt zuständig ist. Es liegt eine Komplexe Heterozygotie vor.
Dieses Gen ist auch beim EEM-Syndrom beteiligt.
Klinisches Erscheinungsbild
Das Haupthaar ist nicht wie gewöhnlich voll und im Wachstum sehr schwach ausgeprägt, am restlichen Körper finden sich normale Haaranlagen. Spätestens zum Schuleintritt ist die Sehbehinderung auch für Außenstehende auffällig.
Die Makuladegeneration zeigt sich in einer langsamen u. U. auch progressiven Verschlechterung der zentralen Sehfähigkeit, später mit Verlust der Lesefähigkeit. Die Betroffenen können sich ansonsten völlig gesund entwickeln und haben eine normale Lebenserwartung.
Diagnostik und Früherkennung
Die Auffälligkeit der Haaranomalie sollte spätestens bei Schuleintritt zu einer Augenhintergrundspiegelung führen, da die üblichen Vorsorgeuntersuchungen nicht zwangsläufig eine Sehbeeinträchtigung erfassen können.
Eine Diagnosesicherung ist nur durch eine molekulargenetische Diagnostik im Rahmen einer humangenetischen Beratung möglich und Voraussetzung für eventuelle zukünftige Therapieoptionen.
Empfohlene Untersuchungsmethode für Verlaufsdiagnostik
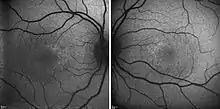
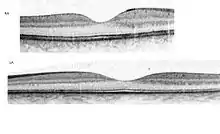
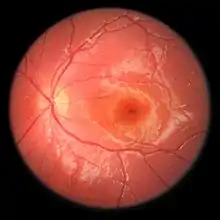
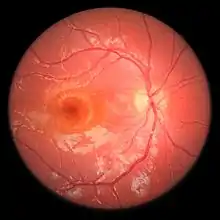
Das Ausmaß der Schädigung der Netzhaut wird durch Fluoreszenzangiografie, Netzhautscan und durch Optische Kohärenztomografie beurteilt, ferner können elektrophysiologische Untersuchung wie das Ganzfeld-Elektroretinogramm (ERG) oder das multifokale Elektroretinogramm (mfERG) genutzt werden.
Differentialdiagnostik
Abzugrenzen sind Haarschaftanomalien im Rahmen einer ektodermalen Dysplasie. Veränderungen im CDH3-Gen können auch beim EEM-Syndrom auftreten.
Therapie
Eine ursächliche Behandlung ist derzeit nicht vorhanden, jedoch bietet mittlerweile eine Vielzahl an Studien in den Bereichen Gentherapie, Exon skipping und CRISPR/Cas zarte Hoffnung am Horizont. Eine genaue Bestimmung der Genmutation durch gesicherte Diagnostik bietet auch weitere potentielle Ansätze jenseits des Genersatzes für eine bestimmte Gruppe – bei einer diagnostizierten sogenannten Nonsens Mutation, eine Mutation bei der durch den Austausch einer einzigen Base in der DNA-Sequenz ein Stop-Codon entsteht. Dadurch kommt es in der Proteinbiosynthese zum vorzeitigen Kettenabbruch. Dadurch resultiert ein verkürztes, funktionsloses bzw. in der Funktion minderwertiges Protein. Translationales Überlesen eines Stop-Codon, die sogenannte „Read-Through-Therapie“ kann durch Verabreichung von bestimmten Stoffen ein Überlesen von Stop-Codons bewirken und somit zu einem brauchbaren Protein führen. Dieser Ansatz ist allerdings nur bei eng begrenzten Mutationen denkbar, die unterschiedliche Erkrankungen verursachen.
Hilfen bei der Krankheitsbewältigung und Lebensplanung
Eine Erkrankung, welche das Augenlicht bedroht und darüber hinaus auch noch eine für Fremde erkennbare Haaranomalie zeigt, verletzt Menschen nicht nur körperlich, sondern immer in ihrer Gesamtheit. So ist es nicht verwunderlich, dass die Diagnoseeröffnung für den betroffenen Patienten einem Schock gleichkommt. Dies gilt bei betroffenen Kindern in gleichem Maße wie für die Eltern und Angehörigen. Man wird mit der Aussage konfrontiert, dass es derzeit keine Behandlungsoption gibt. Nie fühlt man sich in seinem Leben wohl alleiner und im Stich gelassen. Stellen sich die Fragen: „Warum nur ich oder warum mein Kind?“ Es gibt jedoch immer Hoffnung und vor allem bei betroffenen Kindern sollte erstmals eine glückliche Kindheit im Vordergrund stehen. Zu viele Untersuchungen und Arzttermine, kosten Zeit und werden das Problem einer Genmutation nicht praktikabel in wenigen Monaten lösen können. Daher ist es ratsam, dass die Eltern hier einfühlsam mit ihrem Kind umgehen, es aber so erziehen, dass es selbstständig und selbstbewusst ins Teenageralter reifen kann. Ein offenes Umgehen mit der Erkrankung und der Erfahrungsaustausch mit Betroffenen, auch wenn es bei der Seltenheit der Erkrankung unwahrscheinlich ist, jemanden persönlich zu treffen, helfen gemeinsam das Leben zu meistern.
Literatur
- A Rare Syndrome: Hypotrichosis with Juvenile Macular Dystrophy (HJMD)
- CADHERIN 3. In: Online Mendelian Inheritance in Man. (englisch)
- L. Samuelov, E. Sprecher, D. Tsuruta, T. Bíró, J. E. Kloepper, R. Paus: P-cadherin regulates human hair growth and cycling via canonical Wnt signaling and transforming growth factor-β2. In: Journal of Investigative Dermatology. 2012, Band 132, Nr. 10, S. 2332–2341, doi:10.1038/jid.2012.171, PMID 22696062.
- K. Nagel-Wolfrum, F. Möller, I. Penner, U. Wolfrum: Translational read-through as an alternative approach for ocular gene therapy of retinal dystrophies caused by in-frame nonsense mutations. In: Visual neuroscience. Bd. 31, Nr. 4–5, September 2014, S. 309–316, doi:10.1017/S0952523814000194, PMID 24912600 (Review).
- Manuel Jose Justiniano: Micro Perforations Deep Sclerectomy (MPDS). In: Journal of Clinical & Experimental Ophthalmology. 06, 2015, doi:10.4172/2155-9570.S1.022.
- C. Y. Gregory-Evans, X. Wang, K. M. Wasan, J. Zhao, A. L. Metcalfe, K. Gregory-Evans: Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. In: The Journal of clinical investigation. Bd. 124, Nr. 1, Januar 2014, S. 111–116, doi:10.1172/JCI70462, PMID 24355924, PMC 3871240 (freier Volltext).
- N. Schwarz, A.-J. Carr u. a.: Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. In: Human Molecular Genetics. 24, 2015, S. 972, doi:10.1093/hmg/ddu509.
Einzelnachweise
- Hypotrichose - juvenile Makuladegeneration. In: Orphanet (Datenbank für seltene Krankheiten).
- H. Wagner: Maculaaffektion, vergesellschaftet mit Haarabnormitat von Lanugotypus, beide vielleicht angeboren bei zwei Geschwistern. In: Graefes Archiv Klinischer und Experimenteller Ophthalalmologie Bd. 134, 1935, S. 74–81.