Homozygote familiäre Hypercholesterinämie
Die homozygote familiäre Hypercholesterinämie (HoFH) ist eine erbliche, seltene Krankheit.[1] Als Form einer familiären Hypercholesterinämie zählt sie zu den Fettstoffwechselstörungen. Patienten mit HoFH fallen durch eine massive Erhöhung des Low Density Lipoprotein (LDL), einer Cholesterin-Fraktion im Blut, auf.[2][3] Durch die hohen LDL-Cholesterinspiegel kann sich das Lipid langfristig in Haut und Sehnen ablagern, wobei Xanthome (gelbliche, knotenförmige Ablagerung von Lipiden in der Haut) entstehen können. Das Lipid lagert sich auch in den Gefäßwänden ab und verursacht eine früh einsetzende schwere Arteriosklerose mit einer deutlich verkürzten Lebenserwartung.[2][3]
| Klassifikation nach ICD-10 | |
|---|---|
| E78 | Störungen des Lipoproteinstoffwechsels und sonstige Lipidämien |
| E78.0 | Reine Hypercholesterinämie |
| ICD-10 online (WHO-Version 2019) | |
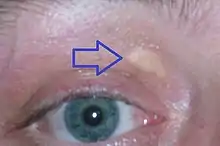
HoFH muss abgegrenzt werden von der sehr viel häufigeren, heterozygoten familiären Hypercholesterinämie (HeFH), die ebenfalls mit einem deutlich erhöhten Risiko für frühzeitige kardiovaskuläre Ereignisse einhergeht, aber weniger dramatisch verläuft.[2][3]
Häufigkeit
In der Literatur wird von einer Krankheitshäufigkeit (Prävalenz) der HoFH von etwa 1:1.000.000 ausgegangen,[4] d. h., es findet sich ein Betroffener auf 1.000.000 Mitglieder einer Bevölkerung. In einigen Populationen ist jedoch von einer höheren Prävalenz auszugehen. Als selten gilt eine Erkrankung, wenn sie mit einer Prävalenz von 1:2000 auftritt, also höchstens bei jedem zweitausendsten Bevölkerungsmitglied.
Zum Vergleich: Die heterozygote Form der familiären Hypercholesterinämie (HeFH) hat eine geschätzte Prävalenz von 1:500.[2] Sie gilt als die häufigste monogenetische Erkrankung. Neuere Populationsuntersuchungen gehen sogar von einer Inzidenz von 1:200 aus.[5]
Ursache
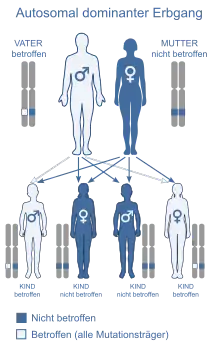
Die familiäre Hypercholesterinämie wird autosomal dominant vererbt.[6] Jedoch können Betroffene bei gleichem Phänotyp Mutationen in verschiedenen Genen aufweisen. Bei homozygoten Individuen werden defekte Allele (Ausprägungsformen eines Gens) von beiden Eltern vererbt. Daher zeigt die Erkrankung eine besonders starke Ausprägung: In der Regel ist die Bindung des LDL-Cholesterins an die Rezeptoren stark beeinträchtigt.[4]
Am häufigsten (85–90 %) finden sich vererbte Gendefekte, welche die Funktion des LDL-Rezeptors beeinträchtigen, der für die Entfernung von LDL-Cholesterin aus dem Blut verantwortlich ist. Dabei kann die Mutation auf über 1.600 Genloci (Genpositionen) liegen.[7]
Andere Ursachen können Mutationen im Gen für das Apolipoprotein B, ein wichtiges Strukturelement des LDL-Cholesterins, oder im Gen für PCSK9 (Proprotein-Convertase Subtilisin/Kexin Typ 9) sein, ein Enzym, das beim LDL-Rezeptor-Abbau eine wichtige Rolle spielt. In seltenen Fällen liegt eine autosomal rezessive Mutation im LDL-Rezeptor Adaptor Protein 1 (LDLRAP1) vor, die in homozygoter Form dem Phänotyp der HoFH entspricht.
Personen mit HoFH können zwei identische Mutationen (klassisch homozygot), zwei unterschiedliche Mutationen im gleichen Gen (compound heterozygot), zwei unterschiedliche Mutationen in zwei verschiedenen Genen (doppelt heterozygot) oder zwei Mutationen im autosomal rezessiven LDL-Rezeptor-AP1-Gen haben (autosomal rezessive Hypercholesterinämie (ARH)).[4][7] Hingegen wird die heterozygote familiäre Hypercholesterinämie durch die Vererbung eines mutierten LDL-Rezeptorallels verursacht.
Symptome und Diagnose
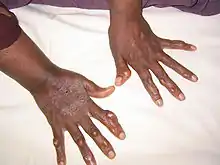

Die klinische Diagnose einer HoFH wird typischerweise aufgrund einer Kombination aus folgenden Kriterien gestellt:
- Massiv erhöhte Gesamtcholesterinwerte von 17 bis 26 mmol/l (650 bis 1000 mg/dl)[8] sowie LDL-Cholesterinwerte > 13 mmol/l (> 500 mg/dl)[4]
- Auftreten von Xanthomen (kutan oder tendinös) bereits in der Kindheit.[2][4]
- Bereits in jungen Jahren Anzeichen einer Arteriosklerose,[2] also Krankheiten und Symptome, die durch Gefäßleiden verursacht sind, z. B. periphere arterielle Verschlusskrankheit, Verengung der Herzkranzgefäße bis zum Herzinfarkt, Schlaganfall.
- Entsprechende Probleme in der blutsverwandten Familie (positive Familienanamnese)[2][3]
Eine genetische Bestätigung der HoFH-Diagnose kann zusätzlich hilfreich sein. Dabei lassen sich funktionale Mutation(en) auf beiden LDL-Rezeptor-Allelen oder Allelen, welche die LDL-Rezeptor-Funktionalität beeinflussen (Apo B, PCSK9 oder ARH), dokumentieren.[4][7] Trotzdem wird in 20 bis 60 Prozent der Fälle trotz klinischer Diagnose mit der gegenwärtigen Gendiagnostik keine Veränderung des Erbgutes (Mutation) gefunden. Es ist davon auszugehen, dass nicht alle HoFH-Patienten über eine Untersuchung der bekannten Gene zweifelsfrei diagnostizierbar sind.[7][9]
Therapie
Therapieziel und -grundsätze
Hauptziel der Therapie der HoFH ist eine dauerhafte Senkung des LDL-Cholesterins im Blut. Die Therapie orientiert sich an den gemeinsamen Leitlinien der European Society of Cardiology (ESC) und der European Atherosclerosis Society (EAS) zur Behandlung von Fettstoffwechselstörungen. Sie definieren für Patienten mit einer familiären Hypercholesterinämie, gleichwohl ob homozygote oder heterozygote Hypercholesterinämie, einen LDL-Cholesterin-Zielwert < 100 mg/dl (Zielwert für Individuen mit hohem kardiovaskulären Risiko) und für Patienten mit einer familiären Hypercholesterinämie und dem Vorliegen einer kardiovaskulären Erkrankung Werte < 70 mg/dl (Zielwert für Individuen mit sehr hohem Risiko).[2]
Die Patienten sollen möglichst in einem spezialisierten Zentrum (Lipidambulanz oder Apheresezentrum) mit lipidsenkenden Medikamenten und LDL-Apherese behandelt werden. Das Behandlungskonzept umfasst zudem eine fettreduzierte, cholesterinarme Ernährung, Nikotinabstinenz und die Empfehlung zur regelmäßigen körperlichen Bewegung.[2][10]
Medikamentöse Therapiemöglichkeiten
HoFH-Patienten sprechen nicht ausreichend auf bisher verfügbare lipidsenkende Pharmakotherapien an. In zwei Studien wurde eine durchschnittliche Senkung des LDL-Cholesterins unter Statinen von sieben Prozent beschrieben.[11] Therapien mit hochdosierten Statinen (Simvastatin, vorwiegend Atorvastatin 80 mg, Rosuvastatin 40 mg), bei 50 % der Patienten in Kombination mit Ezetimib, erreichten unabhängig von der Therapieform eine ca. 26-prozentige Senkung.[12]
Seit 2013 ist in Europa der Wirkstoff Lomitapid in Kombination mit einer fettarmen Diät und anderen lipidsenkenden Arzneimitteln mit oder ohne LDL-Apherese bei erwachsenen Patienten mit homozygoter familiärer Hypercholesterinämie zugelassen.[13] (Für die familiäre Chylomikronämie wurde Lomitapid im Dezember 2010 durch die European Medicines Agency (EMA) der Orphan-Drug-Status gewährt, aber keine Zulassung für diese Indikation erteilt.)[14] Durch die selektive Hemmung des Mikrosomalen Triglycerid-Transfer-Proteins (MTP) kommt es zu einer verminderten Bildung von Lipidkomplexen (Very Low Density Lipoprotein (VLDL) in der Leber, Chylomikronen im Darm). Dadurch wird VLDL nicht mehr von der Leber ins Blut abgegeben bzw. Chylomikronen werden nicht mehr aus dem Darm aufgenommen. Hieraus resultiert eine Senkung der Blutspiegel von VLDL, LDL, Chylomikronen und Apolipoprotein B (Apo B).[15]
Mipomersen ist ein Antisense-Oligonukleotid, welches an die Apo B-kodierende messenger-RNA bindet, dadurch zu einer verminderten Bildung von Apo B in der Leber und entsprechend zu einer Senkung des LDL-Cholesterins führt. Das Medikament wurde in den USA von der Food and Drug Administration (FDA) unter dem Handelsnamen Kynamro® zur Therapie von Patienten mit homozygoter FH unter speziellen Bedingungen zugelassen. Von der Europäischen Kommission wurde die Zulassung 2012 und erneut 2013 abgelehnt, da die aus den Studien ersichtlichen Risiken und Nebenwirkungen (ein hoher Anteil der Patienten hat die Einnahme aufgrund der Nebenwirkungen abgebrochen, Anstieg der Transaminasen, Verfettung der Leber, im Vergleich zur Placebogruppe erhöhte kardiovaskuläre Probleme) die möglichen Vorteile des Wirkstoffes insbesondere im Hinblick auf die notwendige Langzeiteinnahme überwiegen.[16]
Für Patienten mit FH und hohem kardiovaskulärem Risiko stehen seit Ende 2015 zwei vollhumane Antikörper gegen das Protein PCSK9 zur Verfügung, die PCSK9-Hemmer Alirocumab (Handelsname: Praluent) und Evolocumab (Repatha) zur Senkung des LDL-Cholesterins. Ein weiterer PCSK9-Hemmer (Evinacumab) befindet sich in der klinischen Entwicklung. Das Herstellerunternehmen Regeneron Pharmaceuticals (Sanofi) hat von der US-amerikanischen Food and Drug Administration (FDA) den Breakthrough-Therapie-Status für Evinacumab zur Behandlung von Patienten mit homozygoter familiärer Hypercholesterinämie erhalten.[17]
Volanesorsen ist ein Antisense-Hemmer des Apolipoproteins (apo)C-III und wird sowohl bei Patienten mit Hypertriglyzeridämie als auch in der Familiären Chylomikronämie (FCS) untersucht. Die FCS ist ebenfalls eine seltene, autosomal-rezessiv vererbte Störung, die zu den „Orphan Diseases“ (seltene Leiden) gezählt wird und durch eine verminderte oder abwesende Lipoproteinlipaseaktivität (LPL) verursacht wird. FCS zeichnet sich durch eine deutliche Ansammlung von Chylomikronen und extreme Hypertriglyzeridämie aus, normalerweise > 1.000 mg/dl oder 11 mmol/l.
Im April 2021 gab der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittel-Agentur (EMA) eine positive Stellungnahme (positive opinion) ab zur Zulassung von Evinacumab (Handelsname: Evkeeza, Regeneron) zur Behandlung von erwachsenen und jugendlichen Patienten ab 12 Jahren mit homozygoter familiärer Hypercholesterinämie (HoFH). Das Präparat wurde im Rahmen eines beschleunigten Bewertungsprogramms der EMA überprüft. Evinacumab ist ein monoklonaler Antikörper. Er senkt den LDL-C-Cholesterinspiegel (LDL-C) unabhängig von der Anwesenheit des LDL-Rezeptors, indem er die Verarbeitung von Lipoproteinen (VLDL) mit sehr geringer Dichte und die Clearance von VLDL-Resten vor der LDL-Bildung fördert.[18] In der Regel führt ein solches positives Votum innerhalb weniger Wochen zu einer Zulassung in der EU.
LDL-Apherese
Die LDL-Apherese ist ein der Dialyse ähnliches Verfahren, das alle ein bis zwei Wochen durchgeführt wird und das LDL-Cholesterin extrakorporal aus dem Blut herauswäscht. Das wird in Deutschland in ca. 170 Zentren angeboten und die Kosten für diese Therapie werden auf Antrag von den Krankenkassen übernommen. Die Behandlung kann auf Dauer im Mittel zu einer 40- bis 50-prozentigen Reduktion des LDL-Cholesterins führen.[19] Trotz der genannten Reduktion durch Apherese liegen die LDL-Cholesterinwerte bei HoFH Patienten im zeitlichen Durchschnitt noch deutlich über den publizierten Zielwerten von < 100 mg/dl.[20][19] Es wird davon ausgegangen, dass auch mit der LDL-Apherese eine kardiovaskuläre Erkrankung in den meisten Fällen nicht verhindert, sondern nur aufgeschoben werden kann.[10]
Verlauf und Prognose
Bei HoFH-Patienten zeigt sich ein direkter Zusammenhang zwischen der Höhe des LDL-Cholesterin-Spiegels und dem Risiko für kardiovaskuläre Krankheitsereignisse.[21][22] HoFH-Patienten müssen sich daher oft frühzeitig, als Teenager oder junge Erwachsene, Eingriffen am Gefäßsystem (Katheter-Erweiterung, evtl. mit Einsatz einer Gefäßstütze (Stent), Bypass-Operation) unterziehen.[2]
Mit einer maximalen lipidsenkenden medikamentösen Therapie erreichten HoFH-Patienten in einer 2011 veröffentlichten retrospektiven Studie nur ein Durchschnittsalter von 33 Jahren.[12] Eine HoFH-Studie aus dem Jahr 2012 zur zusätzlichen Apherese-Therapie ergab, dass es bei 86 Prozent der Patienten trotz dieser Behandlung zum Fortschreiten der kardiovaskulären Erkrankung kam.[20]
Literatur
- F. J. Raal, R. D. Santos: Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. In: Atherosclerosis. Band 223, Nummer 2, August 2012, S. 262–268, ISSN 1879-1484. doi:10.1016/j.atherosclerosis.2012.02.019. PMID 22398274. (Review).
- S. Walzer, K. Travers u. a.: Homozygous familial hypercholesterolemia (HoFH) in Germany: an epidemiological survey. In: ClinicoEconomics and outcomes research : CEOR. Band 5, 2013, S. 189–192, ISSN 1178-6981. doi:10.2147/CEOR.S43087. PMID 23662069. PMC 3647446 (freier Volltext).
Einzelnachweise
- National Institut of Health (NIH): Liste seltener Erkrankungen (Familial hyperlipo-proteinemia type 1)
- AL. Catapano u. a.: ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). In: European heart journal. Band 37, 2016, S. 2999–3058, doi:10.1093/eurheartj/ehw272.
- P. N. Hopkins, P. P. Toth u. a.: Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. In: Journal of Clinical Lipidology. Band 5, Nummer 3 Suppl, Juni 2011, S. S9–S17, ISSN 1933-2874. doi:10.1016/j.jacl.2011.03.452. PMID 21600530.
- F. J. Raal, R. D. Santos: Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. In: Atherosclerosis. Band 223, Nummer 2, August 2012, S. 262–268, ISSN 1879-1484. doi:10.1016/j.atherosclerosis.2012.02.019. PMID 22398274. (Review).
- B. G. Nordestgaard, M. J. Chapman u. a.: Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. In: European heart journal. Band 34, Nummer 45, Dezember 2013, S. 3478–3490, ISSN 1522-9645. doi:10.1093/eurheartj/eht273. PMID 23956253. PMC 3844152 (freier Volltext).
- W. Siegenthaler, H. E. Blum (Hrsg.): Klinische Pathophysiologie. 9. Auflage. Georg Thieme Verlag, 2006.
- A. C. Goldberg, P. N. Hopkins u. a.: Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. In: Journal of clinical lipidology. Band 5, Nummer 3 Suppl, Juni 2011, S. S1–S8, ISSN 1933-2874. doi:10.1016/j.jacl.2011.04.003. PMID 21600525.
- A. D. Marais: Familial hypercholesterolaemia. In: Clin Biochem Rev. 25(1), 2004, S. 49–86.
- P. J. Talmud, S. Shah u. a.: Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. In: Lancet. Band 381, Nummer 9874, April 2013, S. 1293–1301, ISSN 1474-547X. doi:10.1016/S0140-6736(12)62127-8. PMID 23433573.
- G. R. Thompson, M. Barbir u. a.: Efficacy criteria and cholesterol targets for LDL apheresis. In: Atherosclerosis. Band 208, Nummer 2, Februar 2010, S. 317–321, ISSN 1879-1484. doi:10.1016/j.atherosclerosis.2009.06.010. PMID 19589528. (Review).
- C. Gagné, D. Gaudet, E. Bruckert: Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. In: Circulation. Band 105, Nummer 21, Mai 2002, S. 2469–2475, ISSN 1524-4539. PMID 12034651.
- F. J. Raal, G. J. Pilcher u. a.: Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. In: Circulation. Band 124, Nummer 20, November 2011, S. 2202–2207, ISSN 1524-4539. doi:10.1161/CIRCULATIONAHA.111.042523. PMID 21986285.
- EuropeanMedicines Agency (EMA): Lojuxta / Lomitapide: EPAR summary for the public
- European Medicines Agency (EMA): orphan designation (EU/3/10/823)
- M. M. Hussain, P. Rava u. a.: Multiple functions of microsomal triglyceride transfer protein. In: Nutrition & metabolism. Band 9, 2012, S. 14, ISSN 1743-7075. doi:10.1186/1743-7075-9-14. PMID 22353470. PMC 3337244 (freier Volltext).
- ema.europa.eu Assessment report Kynamro, EMA Committee for Medicinal Products for Human Use (CHMP), July 2013, abgerufen am 3. Februar 2014.
- egeneron Announces Evinacumab has Received FDA Breakthrough Therapy Designation for Homozygous Familial Hypercholesterolemia (HoFH), PM Regeneron vom 6. April 2077, abgerufen am 8. September 2017.
- Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP), 19. bis 22. April 2021, PM EMA vom 23. April 2021, abgerufen am 24. April 2021.
- L. C. Hudgins, B. R. Gordon, T. S. Parker, S. D. Saal, D. M. Levine, A. L. Rubin: LDL Apheresis: an effective and safe treatment for refractory hypercholesterolemia. In: Cardiovasc Drug Rev. 20, 2002, S. 271–280.
- A. Græsdal, M. P. Bogsrud u. a.: Apheresis in homozygous familial hypercholesterolemia: the results of a follow-up of all Norwegian patients with homozygous familial hypercholesterolemia. In: Journal of clinical lipidology. Band 6, Nummer 4, 2012 Jul-Aug, S. 331–339, ISSN 1933-2874. doi:10.1016/j.jacl.2012.03.004. PMID 22836070.
- J. L. Goldstein, H. H. Hobbs u. a.: Familial Hypercholesterolemia. McGraw-Hill, New York 2001.
- S. Moorjani, M. Roy u. a.: Mutations of low-density-lipoprotein-receptor gene, variation in plasma cholesterol, and expression of coronary heart disease in homozygous familial hypercholesterolaemia. In: Lancet. Band 341, Nummer 8856, Mai 1993, S. 1303–1306, ISSN 0140-6736. PMID 8098448.