Epidermolysis bullosa simplex
Die Epidermolysis bullosa simplex (EBS) gehört zu der Krankheitsgruppe der Epidermolysis bullosa und bezeichnet eine Reihe erblicher Erkrankungen mit den Merkmalen einer brüchigen Haut und Bildung von Blasen, spontan oder nach geringem Trauma. Die Schädigung beschränkt sich auf die Epidermis.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| Q81.0 | Epidermolysis bullosa simplex |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
Angaben zur Häufigkeit schwanken von 1 zu 215.000 in den USA bis 1 zu 35.000 in Schottland.
Einteilung
Bislang wurden zahlreiche Formen beschrieben, derzeit betrachtet man 3 – 4 Haupttypen aufgrund deren erheblicher klinischer Überlappung bei Mutationen im gleichen Gen als einheitliche Erkrankung.[2][3]
Haupttypen
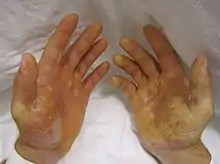
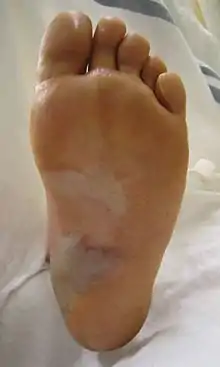
- EBS-WC, Lokalisierter Typ Weber-Cockayne, mildeste und häufigste Form, hauptsächlich Hände und Füße. Mutationen im KRT14-Gen im Chromosom 12 am Genort q21.2 oder am MRT5-Gen im Chromosom 17 am Genort q12-q21.[4][5][6]
- EBS-DM, Herpetiformer Typ Dowling-Meara, generalisiert, schwerste Form, ab Geburt, auch Mundschleimhaut betroffen. Besserung im Alter. Nagelveränderungen, Hyperkeratose an Handflächen und Fußsohlen. Mutationen im KRT5-Gen am Genort 17q12-q21 oder im KRT14-Gen.[7][8][9]
- EBS-K, Generalisierter Typ Köbner, (Generalized epidermolysis bullosa simplex, non-Dowling-Meara type) weniger schwer als Dowling-Meara, Mutationen im KRT14-Gen oder am MRT5-Gen[10][11][6][12]
Seltene Unterformen
- EBS-MD (Epidermolysis bullosa simplex mit Muskeldystrophie; Gliedergürtelmuskeldystrophie – Epidermolysis bullosa simplex), selten, einzige Form ohne Keratinmutation, Muskeldystrophie im Erwachsenenalter, Mutationen im PLEC1-Gen im Chromosom 8 am Genort q24[13][14][6]
- EBS Ogna-Form (Epidermolysis bullosa simplex Ogna), nur in Norwegen, Beginn im Kindesalter, in den Sommermonaten, Mutationen im PLEC1-Gen[15][16][6]
- AR-EBS (Epidermolysis bullosa simplex, autosomal-rezessiv), rezessive Vererbung, Mutationen im KRT14-Gen[17][18]
- EBS-AR BP230 (Epidermolysis bullosa simplex aufgrund von BP230 Mangels), autosomal-rezessiv, Mutationen am DST-Gen im Chromosom 6 am Genort p12.1[19][20]
- EBS-AR exophilin 5 (Epidermolysis bullosa simplex due to exophilin 5 deficiency), autosomal rezessiv, Mutationen im EXPH5-Gen im Chromosom 11 am Genort q22.3[21][22]
- LAEB (Letale akantholytische Epidermolysis bullosa), autosomal-rezessiv, Blasen suprabasal, Mutationen im DSP-Gen im Chromosom 6 am Genort p24.3, das für Desmoplakin kodiert[23][24]
- EBS-PD(Epidermolysis bullosa simplex durch Plakophilin-Mangel; Ektodermale Dysplasie-Hautfragilität-Syndrom; McGrath-Syndrom), autosomal-rezessiv, Blasen suprabasal, Mutationen im PKP1-Gen im Chromosom 1 an q32, das für Plakophilin-1 kodiert[25][26]
- EBS-MP (Epidermolysis bullosa simplex fleckiger Hyperpigmentierung, englisch mottled pigmentation) mit gesprenkelter Pigmentierung, dunkle Flecken an Rumpf, Armen und Beinen, im Erwachsenenalter abnehmend, mit milder Nageldystrophie und fokalem palmo-plantarem Keratoderma, Mutationen im KRT5-Gen[27][28][29][2][6]
- EBS-migr (Epidermolysis bullosa simplex mit ringförmigem Erythema migrans|englisch EPIDERMOLYSIS BULLOSA SIMPLEX WITH MIGRATORY CIRCINATE ERYTHEMA), ab Geburt, gürtelartiges Erythem mit zahlreichen Blasen im Randbereich, Mutationen im KRT5-Gen[30][31]
- EBSS (Epidermolysis bullosa simplex superficialis), Läsionen suprabasal ohne Blasenbildung[32][33]
- Epidermolysis bullosa simplex mit Anodontie/Hypodontie(Gamborg-Nielsen-Syndrom; Kallin-Syndrom)[34]
- EBS-PA, autosomal-rezessiv, Kombination mit Pylorusatresie, Mutationen am PLEC1-Gen[35][36]
Klinik
Die Vererbung erfolgt in der Regel autosomal-dominant, die seltenen Unterformen AR-EBS, EBS-AR BP230, EBS-AR exophilin 5, LAEB, EBS-PD, EBS-PA werden autosomal-rezessiv vererbt.[1]
Gemeinsame Merkmale sind:[1][6]
- Krankheitsbeginn während oder kurz nach der Geburt, aber auch im Kindes- oder frühen Erwachsenenalter
- Auftreten von Blasen und Erosionen
Lokalisation der Blasen in der Basalschicht der Haut, bei wenigen Formen (dort angegeben) suprabasal.
Hinzu können Nagelablösung oder Nageldystrophie sowie Keratoderma der Handflächen und Fußsohlen treten. Narbige Veränderungen entwickeln sich meist nicht oder nur geringgradig. Außerhalb der Haut kommen Blasenbildungen in der Mundhöhle vor.
Diagnostik
Die Diagnose basiert auf der Bestimmung der Epidermisschicht, in der nach leichtem Zug an der Haut die Blasen entstehen, in einer Hautbiopsie mithilfe von Immunfluorimetrisches Antigen-Mapping und Transmissions-Elektronenmikroskopie, der Art der Vererbung und der Klinik.[1]
Differentialdiagnostik
Lediglich in der Neugeborenenzeit besteht ein Abgrenzungsbedarf gegenüber:[1]
- Herpes simplex
- Kongenitale Aplasia cutis
- Pemphigus acutus neonatorum
- Neonataler Herpes gestationis
- Lyell-Syndrom
- Incontinentia pigmenti
- Epidermolytische Ichthyose
- Lineare IgA-Dermatose
- Bullöses Pemphigoid
- Impetigo contagiosa
Therapie
Die Behandlung besteht im Vermeiden der Blasenbildung durch Polsterung der Haut, Vermeidung von Traumata.
Siehe auch
- Mutationen im KRT14-Gen mit Veränderungen am Keratin 14 liegen auch bei der Dermatopathia pigmentosa reticularis und dem Naegeli-Syndrom vor.[37]
- Mutationen im KRT5-Gen mit Veränderungen am Keratin 5 liegen auch bei der Dowling-Degos-Krankheit Typ 1 vor.[38]
Einzelnachweise
- Epidermolysis bullosa simplex. In: Orphanet (Datenbank für seltene Krankheiten).
- Medline Plus
- Altmeyers Enzyklopädie Epidermolysis bullosa simplex
- Epidermolysis bullosa simplex, lokalisierte. In: Orphanet (Datenbank für seltene Krankheiten).
- Enzyklopädie Dermatologie Epidermolysis bullosa simplex Weber-Cockayne
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Epidermolysis bullosa simplex Dowling-Meara
- Epidermolysis bullosa simplex Typ Dowling-Meara. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa herpetiformis. In: Online Mendelian Inheritance in Man. (englisch)
- Generalized epidermolysis bullosa simplex. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex Köbner
- epidermolysis bullosa simplex, non-Dowling-Meara type 11425. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex mit Muskeldystrophie. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex with muscular dystrophy. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex Typ Ogna. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex of Ogna. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex, recessive 1. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex, autosomal-rezessive. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex due to BP230 deficiency. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex, autosomal recessive 2. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa, nonspecific, autosomal recessive. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex due to exophilin 5 deficiency. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa, akantholytische letale. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa, lethal acantholytic. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex durch Plakophilin-Mangel. In: Orphanet (Datenbank für seltene Krankheiten).
- Ectodermal dysplasia/skin fragility syndrome. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex-MP. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex mit gesprenkelter Pigmentierung. In: Orphanet (Datenbank für seltene Krankheiten).
- Enzyklopädie Dermatologie Epidermolysis bullosa simplex mit fleckiger Hyperpigmentierung
- Epidermolysis bullosa simplex mit ringförmigem Erythema migrans. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermylysis bullosa simplex-MCR. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex superficialis. In: Orphanet (Datenbank für seltene Krankheiten).
- EBSS. In: Online Mendelian Inheritance in Man. (englisch)
- Epidermolysis bullosa simplex mit Anodontie/Hypodontie. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex mit Pylorusatresie. In: Orphanet (Datenbank für seltene Krankheiten).
- Epidermolysis bullosa simplex with pyloric atresia. In: Online Mendelian Inheritance in Man. (englisch)
- KRT14
- KRT5