Eosinophile Granulomatose mit Polyangiitis
Die eosinophile Granulomatose mit Polyangiitis (EGPA) (älter auch Churg-Strauss-Syndrom) ist eine sehr seltene granulomatöse („körnchenbildende“) Entzündung von Blutgefäßen, bei der das betroffene Gewebe von bestimmten Entzündungszellen, den eosinophilen Granulozyten, infiltriert („durchwandert“) wird. Es ist zum Großteil nachweislich verknüpft mit der Konzentrationserhöhung einer bestimmten Unterklasse von Antikörpern (ANCA (s. u.)) und betrifft vor allem kleinere und mittlere Arterien der Lunge und anderer Organe. Durch die Entzündungsreaktion werden Gewebe und Organe geschädigt. Die Namensgebung stammt von den Pathologen Jacob Churg und Lotte Strauss, die die Krankheit 1951[1] zuerst beschrieben.
| Klassifikation nach ICD-10 | |
|---|---|
| M30.1 | Panarteriitis mit Lungenbeteiligung Allergische Granulomatose Churg-Strauss-Granulomatose |
| ICD-10 online (WHO-Version 2019) | |
Pathogenese
Die Ursache dieser auch als eine „allergische Angiitis und Granulomatose“ bezeichneten Erkrankung ist weitestgehend unbekannt. Eine ANCA-assoziierte autoimmunologische Komponente wird vermutet. So erfolgt auch die Behandlung immunsuppressiv. Bei Erkrankten sind die Zytokine Interleukin-17A und Interleukin-23 im Serum erhöht.[2]
Prävalenz
Pro Jahr treten etwa 1–3 Fälle / 1 Million Menschen auf. Das Durchschnittsalter der Erstdiagnose sind 48 Jahre. Es sind mehr Frauen als Männer betroffen.[3]
Pathologie
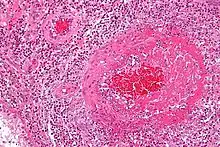
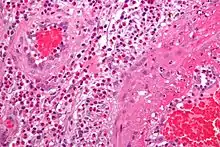
Feingeweblich zeigt sich eine starke Vermehrung eosinophiler Granulozyten im Gewebe (Gewebseosinophilie), mit Befall vor allem der kleinen Blutgefäße (mit Zerstörung, Blutgerinnselbildung und daraus resultierenden Infarkten). Daneben kann die Entzündung auch direkt auf verschiedene Organe übergreifen, z. B. auf das Herz mit der Folge einer eosinophilen Herzmuskelentzündung.
Symptome
Die EGPA hat drei verschiedene klinische Phasen:
- allergischer Schnupfen und Asthma bronchiale
- eosinophile Entzündung von Lunge und Verdauungstrakt
- systemische Gefäßentzündung (Vaskulitis) mit granulomatöser Entzündung
Die vaskulitische Phase beginnt in der Regel etwa drei Jahre nach Anfang der ersten Phase, kann aber auch um Jahrzehnte verzögert auftreten. Außerdem tritt ein Befall von Lunge, Herz, Niere, Haut und peripherem Nervensystem auf. Folgende Symptome sind typisch für die EGPA:
- Allgemeinsymptome (Müdigkeit, grippeartige Symptome wie Fieber und Muskelschmerzen, allgemeines Unwohlsein, Gewichtsverlust in 70 Prozent der Fälle)
- Asthma (97 Prozent der Fälle)
- Nasennebenhöhlenentzündung (61 Prozent der Fälle)
- allergischer Schnupfen
- Gelenkschmerzen
- Hautmanifestationen (Purpura, Hautknötchen, Nesselsucht)
- Herzsymptome, welche auf Herzinfarkt und Herzmuskelentzündung schließen lassen.
- Nervenentzündung
- durch Nierenbeteiligung Bluthochdruck und Nierenversagen
- Symptome, den Magen-Darm-Trakt betreffend (Blutungen, „Blinddarmentzündung“, Bauchspeicheldrüsenentzündung, Magen-Darm-Beschwerden)
Im Labor findet man eine Eosinophilie und Blutarmut sowie Entzündungszeichen (erhöhte Blutsenkungsgeschwindigkeit und erhöhtes CRP). Weiterhin findet man antineutrophile cytoplasmatische Antikörper (p-ANCA), erhöhte IgE-Spiegel und einen erhöhten Rheumafaktor.
Im Röntgenbild des Brustkorbs findet man bei 25–75 Prozent der Fälle beidseitige, ungleichmäßig verteilte Verschattungen in der Lunge.
Beweisend für die EGPA ist die Biopsie aus einem betroffenen Organ oder, wenn dies nicht vorhanden ist, die Biopsie aus Muskel- oder Nervengewebe (dann meist aus dem Nervus suralis).
EGPA ist wie alle Vaskulitiden vielschichtig und schwierig in der Diagnose. Die oben angegebenen Allgemeinsymptome brauchen nicht sämtlich aufzutreten. Bei einem unklaren Krankheitsbild ist es aber immer sinnvoll, auch eine Vaskulitis als Auslöser mit einzubeziehen.
Differentialdiagnose
Abzugrenzen ist das Hypereosinophilie-Syndrom sowie die klinisch ähnliche Granulomatose mit Polyangiitis.
Therapie
Unbehandelt wird das 5-Jahres-Überleben nur mit ca. 25 % angegeben. Eine Studie berichtete von einer Überlebenswahrscheinlichkeit von 72 % in 6½ Jahren bei Patienten die behandelt wurden. Die häufigste Todesursache sind dabei Herz-Kreislauferkrankungen.[3]
In vielen Fällen reicht eine Therapie mit Prednison aus, um die Symptome zu kontrollieren. Bei schwerwiegenden Organbeteiligungen ist eine Kombination mit Chemotherapeutika wie Cyclophosphamid Mittel der Wahl.[3] Andere Möglichkeiten der Behandlung sind der Einsatz von Methotrexat, Interferon-α, Immunglobulinen oder der Plasmapherese. Am 12. Dezember 2017 erhielt der Anti-Interleukin-5-Antikörper Mepolizumab die FDA-Zulassung zur Behandlung erwachsener Patienten mit EGPA.[4]
Nomenklatur
Die EGPA gehört zum Formenkreis der systemischen, nekrotisierenden entzündlichen Erkrankungen der kleinen und mittleren Arterien. Von der klassischen Periarteriitis nodosa (Panarteriitis) ausgehend, über die Kussmaul und Maier 1866 berichtet hatten, beschrieb Wegener 1939 eine granulomatöse Vaskulitis der oberen Atemwege als eigenes Syndrom. Churg und Strauss stellten 1951 ein weiteres Krankheitsbild mit Asthma bronchiale und peripherer Eosinophilie vor. Sie nannten diese Variante ursprünglich „allergische Granulomatose“ und „allergische Angiitis“.
Literatur
- J. A. Eustace, T. Nadasdy, M. Choi: The Churg Strauss Syndrome. In: J Am Soc Nephrol. Band 10, Nr. 9, September 1999, S. 2048–2055. Review. PMID 10477159 (englisch)
- Franz Hrska, Wolfgang Graninger, Michael Frass: Systemerkrankungen. In: Anästhesiologie Intensivmedizin Notfallmedizin Schmerztherapie. Band 38, Nr. 11, (November) 2003, S. 719–740, hier: S. 728 f. (Churg-Strauss-Syndrom) und 735.
Weblinks
- Rheuma Online; abgerufen am 25. Juli 2012
- ANCA-assoziierte Vaskulitis, Deutsche Nierenstiftung
Einzelnachweise
- J. Churg, L. Strauss: Allergic Granulomatosis, Allergic Angiitis, and Periarteritis nodosa. In: Am J Pathol. Band 27, 1951, S. 277–301. PMID 14819261 (englisch).
- E. Nogueira, S. Hamour, D. Sawant u. a.: Serum IL-17 and IL-23 Levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. In: Nephrol Dial Transplant. 2010; 25, S. 2209–2217. PMID 20100727
- Kasper, Fauci: Harrison’s Principles of Internal Medicine. 19. Auflage. Band 2. McGraw-Hi11 Education, New York 2015, ISBN 978-0-07-180215-4, S. 2186–2187.
- https://arznei-news.de/mepolizumab/