Cori-Krankheit
Die Cori-Krankheit (auch Forbes-Krankheit, Morbus Cori, Typ III-Glykogen-Speicherkrankheit oder Amylo-1,6-Glucosidase-Mangel) ist eine autosomal-rezessiv vererbte Krankheit, die den Glykogenhaushalt beeinträchtigt und nach 1953 von Barbara Illingworth, Forbes, Carl und Gerty Cori entdeckt und beschrieben wurde. Coris Krankheit wird von einer Mutation auf dem Locus 1p21.2 verursacht.
| Klassifikation nach ICD-10 | |
|---|---|
| E74.0 | Glykogenspeicherkrankheit [Glykogenose] Cori-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
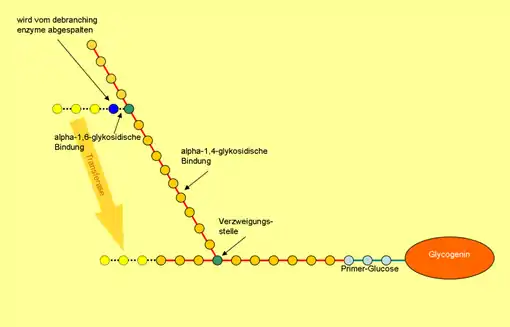
Glykogen ist eine Art Zucker, ein hoch verzweigtes Polymer, das als Energiespeicher in der Leber und in den Muskeln vorherrscht. Bei der Cori-Krankheit kann es nicht mehr korrekt verarbeitet werden. Ein defektes Enzym, das Glykogen-Debranching-Enzym, kann die Verzweigungen des Glykogens nicht mehr abbauen. Die Folgen sind:
- Hypoglykämie schon im Embryonalstadium, da die Krankheit genetisch veranlagt ist – doch die Unterzuckerung äußert sich oft erst im Alter von drei bis vier Monaten, wenn die Brusternährung eingestellt wird
- gehemmtes Wachstum
- üblicherweise eine vergrößerte Leber
- später sind auch die Muskeln betroffen (Hypotonie, Herzmuskelerkrankungen)
Die Leber beginnt sich zu normalisieren, sobald der Patient erwachsen wird; doch wenige Betroffene entwickeln eine Leberzirrhose im Erwachsenenalter. Die Lebersymptomatik kann durchaus so mild sein, dass die Krankheit erst im Erwachsenenalter durch die neuromuskulären Symptome erkannt wird.
Die Behandlung besteht aus einer speziellen Ernährung. Patienten sollten längere Fastenperioden vermeiden, und häufige kleinere Mahlzeiten einnehmen, dabei aber Einfachzucker und andere schnell resorbierbare Kohlenhydrate in der Menge je Mahlzeit begrenzen. Eine wichtige Rolle in der Ernährungstherapie spielt ungekochte Maisstärke, da diese besonders langsam verdaut wird und dadurch der Blutzuckerspiegel über mehrere Stunden unterstützt werden kann. Eine sehr proteinreiche Diät ist angezeigt, gegebenenfalls mit Supplementation von Proteinen, da durch die Gluconeogenese Aminosäuren in Glucose umgewandelt werden können.[1]
Häufigkeit
- In den USA und in Europa 1 Patient pro 83.000 bis 100.000 Neugeborene
- Nordamerikanische Inuit und sephardische Juden nordafrikanischer Abstammung ca. 1:5000 (Heirat oft innerhalb der eigenen Subpopulation, was Homozygotie pathogener Allele fördert)
- Färöer-Inseln: ca. 1:3600 (Gründereffekt bzw. genetischer Flaschenhals)
Literatur
- A. Dagli, C. P. Sentner, D. A. Weinstein: Glycogen Storage Disease Type III. In: GeneReviews. University of Washington, Seattle, Seattle (WA) 29. Dezember 2016, PMID 20301788 (nih.gov [abgerufen am 19. Januar 2018]).