Angeborene Hypothyreose
Die angeborene (congenitale, auch connatale) Hypothyreose stellt eine besondere Form der Schilddrüsenunterfunktion dar. Sie betrifft die Patienten in einer Entwicklungsphase, in der ihr gesamtes Nervensystem besonders empfindlich ist und durch das Fehlen von Schilddrüsenhormonen nachhaltig geschädigt werden kann. Wird die Störung nicht unmittelbar nach der Geburt erkannt und behandelt, führt sie zum Vollbild des Kretinismus.
| Klassifikation nach ICD-10 | |
|---|---|
| E03.0 | Angeborene Hypothyreose mit diffuser Struma |
| E03.1 | Angeborene Hypothyreose ohne Struma |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
Der Screening-Report der Deutschen Gesellschaft für Neugeborenenscreening (DGNS) weist für das Jahr 2004 eine Gesamtprävalenz von 1:3270 Neugeborenen auf (Prävalenz 2013: 1:3233). Innerhalb der einzelnen Screening-Zentren schwankt die Häufigkeit zwischen 1:2143 und 1:5239.[1] Diese Zahlen entsprechen etwa denjenigen, die für Nordamerika angegeben werden, wo eine Häufigkeit von etwa 1:3000 bis 1:4000 Neugeborenen mit etwas größerer Häufigkeit unter den spanischstämmigen und geringeren Werten bei afroamerikanischen Kindern berichtet wird.[2] Damit ist die angeborene Schilddrüsenunterfunktion ungefähr doppelt so häufig wie die Phenylketonurie, die erste angeborene Stoffwechselerkrankung, für die ein Screening eingeführt wurde. Mädchen sind etwa doppelt so oft betroffen wie Jungen.[2]
Eine angeborene Hypothyreose kann im Rahmen seltener Syndrome wesentliches Merkmal sein, so bei dem Hirn-Lunge-Schilddrüsen-Syndrom.
Ursachen
Je nach Ursache kann unterschieden werden zwischen:
- Gestörte Organentwicklung
- In den meisten Fällen wird die Unterfunktion der Schilddrüse durch eine Störung in ihrer Organentwicklung während der Embryonalentwicklung (Agenesie oder Dysgenesie) verursacht. Dabei entsteht gar kein oder zu wenig funktionsfähiges Schilddrüsengewebe. Der Grund für diese Fehlanlagen ist nicht bekannt. Lediglich in seltenen Einzelfällen konnten Mutationen in Genen nachgewiesen werden, die für die Entwicklung der Schilddrüse eine Rolle spielen, darunter im FOXE1-Gen, das für den Thyroidalen Transkriptionsfaktor 2 (TTF-2) kodiert, und im PAX8-Gen, das für das Paired-Box-Protein 8 (PAX-8) kodiert.[3]
- Gestörte Schilddrüsenfunktion
- Daneben gibt es Störungen der Hormonbildung in einer ansonsten regelrecht angelegten Schilddrüse. Angeborene Störungen der Thyroxin-Synthese folgen einem autosomal-rezessiven Erbgang, Defekte im Rezeptor für das Schilddrüsenhormon werden autosomal-dominant vererbt. Insgesamt treten etwa 85 % aller angeborenen Schilddrüsenunterfunktionen sporadisch auf, die übrigen 15 % sind erblich bedingt.[3]
- Transitorische Hypothyreose
- Seltener ist die Unterfunktion vorübergehender Natur. Dies ist der Fall, wenn sie durch Übertragung von mütterlichen Medikamenten oder blockierenden Antikörpern über den Mutterkuchen auf das Kind, durch Jodmangel oder exzessive Jodbelastung verursacht wird.
- Zentrale Hypothyreose
- Störungen der Hypothalamus-Hypophysen-Achse – sogenannte zentrale, sekundäre oder tertiäre Unterfunktionen – mit eingeschränkter Produktion von Thyreotropin (TSH) oder Thyrotropin Releasing Hormone (TRH) sind extrem selten, beispielsweise beim IGSF1-Mangelsyndrom, entgehen aber den üblichen Screeninguntersuchungen.[2]
Klinische Erscheinungen
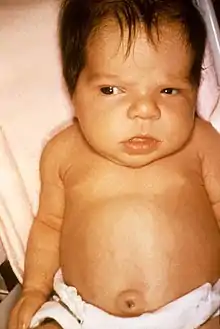

Obwohl das Schilddrüsenhormon für die Entwicklung fast aller Organsysteme von außerordentlicher Bedeutung ist, sind die meisten Kinder bei der Geburt zunächst unauffällig. Durch mütterliches Schilddrüsenhormon, das über die Plazenta übertragen wird, sind selbst diejenigen Feten, denen eine Schilddrüse vollständig fehlt, zumindest teilweise geschützt. Im Nabelschnurblut dieser Kinder beträgt die Konzentration an Thyroxin (T4) etwa ein Drittel bis die Hälfte derjenigen von gesunden Kindern. Die etwa 5 % am schwersten betroffenen Kinder können schon früh mit großen Fontanellen, weit klaffenden Schädelnähten, großer Zunge, aufgetriebenen Bauchdecken und Nabelbruch auffallen.[3] Mit zunehmendem Abbau des mütterlichen Thyroxins treten weitere Symptome hinzu. Die Säuglinge trinken schlecht, bekommen eine Verstopfung, sind teilnahmslos und schlafen viel. Oft müssen sie zu den Mahlzeiten geweckt werden. Die Stimme kann rau und heiser sein, die Haut fühlt sich kühl an, die Spannung der Muskulatur ist schlaff (Hypotonie) und die Reflexe sind schwach. Eine Neugeborenengelbsucht (Icterus neonatorum) besteht aufgrund der verzögerten Reifung der Leber möglicherweise verlängert (Icterus prolongatus). Eine Vergrößerung der Schilddrüse (Kropf, Struma) findet man in etwa 5–10 % der Kinder, zumeist bei angeborener Störung der Thyroxin-Synthese.[3]
Wird die Erkrankung nicht erkannt und behandelt, zeigt sich ab dem zweiten bis dritten Lebensmonat eine Verzögerung des Wachstums. Die Minderung der Intelligenz nimmt ebenfalls zu, je später die Behandlung einsetzt. Beginnt sie in den ersten drei Monaten, liegt der mittlere Intelligenzquotient bei 89 (Spannweite 64–107), bei Beginn zwischen dem vierten und sechsten Lebensmonat bei 71 (Spannweite 35–96) und setzt sie erst nach dem ersten Lebenshalbjahr ein bei 54 (Spannweite 25–80).[3] Andere Langzeitfolgen sind Störungen in der grob- und feinmotorischen Koordination, Gleichgewichtsstörungen (Ataxien), Muskelschwäche und Spastizität, Sprachstörungen, Aufmerksamkeitsstörungen und Schielen.
Untersuchungsmethoden
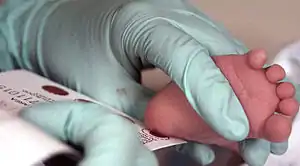
Da eine frühe Diagnosestellung für eine möglichst normale Entwicklung der Kinder so enorm wichtig ist, wird die Diagnose in den meisten entwickelten Ländern über eine Reihenuntersuchung im Neugeborenenalter (Neugeborenenscreening) gestellt. Dabei wird in Deutschland der Gehalt an Thyreotropin (TSH), welches bei fehlender Produktion von Schilddrüsenhormon deutlich ansteigt, in sogenanntem Trockenblut bestimmt. Allerdings werden dabei die seltenen Fälle einer zentralen bedingten sekundären oder tertiären Hypothyreose übersehen.[4] In den Vereinigten Staaten von Amerika messen die meisten Screening-Programme zunächst die T4-Konzentration und nur bei Werten unter der zehnten Perzentile zusätzlich das Thyreotropin.[3] Die Probenentnahme soll unbedingt schon am dritten Lebenstag erfolgen. Wird das Neugeborene schon vorher entlassen oder verlegt, kann sie auch schon früher erfolgen, muss dann aber in der Regel wiederholt werden.[4] Im positiven Fall muss dann umgehend ein Bestätigungstest mit einer Bestimmung von Thyreotropin, freiem T4 und freiem T3 erfolgen. Bestätigt sich der Befund einer Schilddrüsenunterfunktion, können weitere Untersuchungen zur Abklärung der Ursache durchgeführt werden. Dazu gehören neben der Bestimmung von Thyreoglobulin zum Nachweis von Schilddrüsengewebe und einer Hormonbildungsstörung vor allem bildgebende Maßnahmen wie Sonografie oder Schilddrüsenszintigrafie. Da die Ergebnisse dieser Untersuchungen aber keinen Einfluss auf die Behandlung haben, sind sie nicht zwingend notwendig.[3] Wenn es Hinweise auf eine mütterliche Schilddrüsenerkrankung gibt, kann die Bestimmung von Schilddrüsenautoantikörpern bei Mutter und Kind jene Formen aufdecken, die voraussichtlich vorübergehender Natur sind. Besteht der Verdacht auf einen Jod-Exzess oder einen Jodmangel als Ursache der Hypothyreose, kann dieser durch eine Bestimmung der Jodausscheidung mit dem Urin bestätigt werden.[3]
Behandlung
Wegen der möglicherweise nicht mehr rückgängig zu machenden Schädigung des Nervensystems muss die Behandlung einer angeborenen Hypothyreose so früh wie möglich erfolgen. Sie darf nicht unnötig durch Abwarten von Untersuchungsergebnissen verzögert werden und soll unmittelbar nach der Blutentnahme für die Bestätigungsdiagnostik bei positivem Screeningbefund erfolgen.[2] Ziel der Behandlung ist eine möglichst rasche Normalisierung des T4-Gehalts mit einer nachfolgenden Normalisierung des TSH-Gehalts des Bluts. Hierzu wird das Hormondefizit durch Gaben von Levothyroxin in einer relativ hohen anfänglichen Dosierung ausgeglichen. Nach zwei Wochen kann die Dosis je nach Ergebnis einer Kontrolle der Werte für TSH und T4 im Blut reduziert werden. Entsprechend dem Wachstum des Kindes muss sie im weiteren Leben dem erhöhten Bedarf angepasst werden. Bestehen Zweifel an der Diagnose einer dauerhaften Schilddrüsenunterfunktion, kann ein Auslassversuch frühestens nach dem ersten Geburtstag, nach amerikanischen Quellen besser erst nach dem dritten Geburtstag gemacht werden.[2] Andernfalls muss die Substitution lebenslang erfolgen. Ist ein Jodmangel als Ursache der Unterfunktion der Schilddrüse nachgewiesen, wird dieser mit einer Jodsubstitution behandelt.
Heilungsaussicht
In Abhängigkeit vom Zeitpunkt der Diagnosestellung ist die Prognose für die betroffenen Kinder sehr gut. Wird die Unterfunktion schon innerhalb der ersten beiden Lebenswochen erkannt und konsequent behandelt, finden sich im Erwachsenenalter nur geringe Unterschiede in Intelligenzniveau, Schulleistungen und neurophysiologischen Tests gegenüber gesunden Klassenkameraden oder Geschwistern.[2] Beginnt die Behandlung jedoch erst später, kann sich zwar die körperliche Entwicklung noch normalisieren, die Intelligenz bleibt jedoch eingeschränkt.
Einzelnachweise
- Screeningreport 2004 der Deutschen Gesellschaft für Neugeborenenscreening (Memento vom 2. September 2007 im Internet Archive)
- American Academy of Pediatrics, American Thyroid Association, Lawson Wilkins Pediatric Endocrine Society: Update of Newborn Screening an Therapy for Congenital Hypthyroidism. In: Pediatrics. 2006, 117, S. 2290–2303.
- C. I. Kaye und the Committee on Genetics: Newborn Screening Fact Sheet. In: Pediatrics. 2006; 118, S. e934–e963, PMID 16950973.
- Leitlinie der GNPI (Memento vom 24. Februar 2008 im Internet Archive)