Undine-Syndrom
Das Undine-Syndrom oder kongenitale zentrale Hypoventilationssyndrom (früher auch Undine-Fluch-Syndrom) ist eine seltene angeborene Erkrankung des zentralen Nervensystems, dessen Hauptsymptom eine Störung der Atemregulation ist. Aus dem Englischen übertragen ist die Abkürzung CCHS (Congenital Central hypoventilation syndrome) auch im deutschsprachigen Bereich gebräuchlich.
| Klassifikation nach ICD-10 | |
|---|---|
| G47.3 | (zentrale) Schlafapnoe |
| ICD-10 online (WHO-Version 2019) | |
Das Syndrom umfasst neben der Atmungsstörung auch weitere Fehlregulationen autonomer Funktionen wie Über- oder Untertemperatur, Herzrhythmusstörungen, Schluckstörungen oder selten Tumoren. Das gleichzeitige Vorliegen eines CCHS und eines Morbus Hirschsprung wird auch als Haddad-Syndrom[1][2] bezeichnet.
Die Schlafmedizin spezifiziert diese Schlafstörung als Schlafbezogene Atmungsstörung und teilt sie nach ICD-10-GM in G47.35 (Angeborenes zentrales alveoläres Hypoventilationssyndrom[3]) oder als Schlafapnoe in G47.32 (Kongenitales zentral-alveoläres Hypoventilations-Syndrom[4]) ein.
In der Internationalen Klassifikation der Schlafstörungen International Classification of Sleep Disorders ICDS-3[5] werden die Diagnosekriterien festgelegt. Diese sind das Vorliegen einer schlafbezogenen Hypoventilation und das Vorhandensein einer Mutation im PHOX2B-Gen.
Verbreitung / Inzidenz
Betroffen ist etwa einer von 180.000 Lebendgeborenen.[6]
Ursache
Das PHOX2B-Gen
Das die Krankheit definierende Gen ist das PHOX2B-Gen, welches auf dem kurzen Arm des Chromosoms Nummer 4 liegt.[7] Es besteht aus 3 Exons, welche Informationen für den Aufbau des PHOX2B-Proteins (Eiweiß) in der DNA kodieren. Im Exon 3 liegt eine Region, die für eine Wiederholung eines Einbaus der Aminosäure Alanin in das Protein kodiert. Im Normalfall werden in dieser Genregion 20 Alanine hintereinander kodiert. Daher wird die Stelle im Gen Polyalaninrepeat, (wörtlich: Vielfachalaninwiederholung) oder kurz PAR, genannt.
Mutationen im PHOX2B-Gen
Bei ca. 80 % der CCHS-Patienten liegt eine Verlängerung des PARs vor. Ab einer Verlängerung von 4 Alaninen tritt die Erkrankung auf. Die längste Verlängerung des PARs ist 33 Alanine.[8] Diese Art von Gen-Veränderung wird als Polyalaninrepeat-Mutation bezeichnet und mit PARM abgekürzt. Die in der Normalbevölkerung vorkommende Allelkombination wird bezüglich des PARs als 20/20 beschrieben. Beide PHOX2B-Gene enthalten einen PAR von 20 Alaninen. Bei PARMs, die das CCHS verursachen können, lauten diese 20/24, 20/25, 20/26 ... bis 20/33. Sehr selten kann auch eine Verkürzung des PARs ein CCHS verursachen.
Neben den PARMs kommen auch Frameshift-, Punkt- oder Missense-Mutationen vor. Diese Genveränderungen liegen bei ungefähr 20 % der Patienten vor.
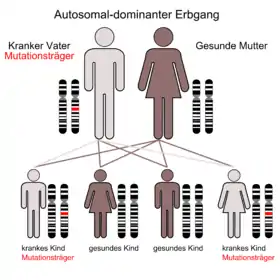
Zumeist handelt es sich um Neumutationen, welche bei den CCHS-Erkrankten festgestellt werden. Die Eltern sind in dem Fall weder erkrankt, noch tragen sie das veränderte Gen in sich. Die Krankheit kann jedoch auf Kinder vererbt werden, wenn ein Elternteil selbst das CCHS hat oder eine Mutation für das betreffende Gen in den Keimdrüsen trägt. Letzteres kann ohne jedes Symptom vorkommen und wird als Genetisches Mosaik bezeichnet.
Der Erbgang der Erkrankung ist autosomal dominant. Damit wird die Erkrankung mit einer statistischen Wahrscheinlichkeit von 50 % auf die Kinder übertragen, wenn ein Elternteil selbst betroffen ist.
Symptome
Hypoventilation
Dabei handelt es sich um eine Störung der zentralen CO2-Chemorezeptorsensitivität (verminderte CO2-Antwort). Die Atemantwort eines Menschen mit CCHS auf eine Hyperkapnie (Kohlendioxidanstieg) im Blut oder eine Hypoxie (niedrige Sauerstoffsättigung) ist typischerweise im Schlaf, bei vielen aber auch im Wachzustand, eingeschränkt. Bei Neugeborenen fallen die fehlende Spontanatmung oder eine unzureichende, oberflächliche Atmung auf. Dies ist im Schlaf stärker ausgeprägt als im Wachzustand und kann von Episoden mit normaler Atmung unterbrochen werden. Während der Hypoventilationsphasen und besonders im Tiefschlaf kann es dabei zu lebensbedrohlichen Zuständen kommen.[3]
Morbus Hirschsprung
Etwa 25 % der Undine-Kinder leiden zusätzlich an Morbus Hirschsprung, einer angeborenen Fehlbildung des Darmes, dessen Hauptsymptom eine Verstopfung (Obstipation) bzw. ein chronischer Stuhlverhalt ist. Verantwortlich für die Symptome ist eine Engstellung des Darmes und unter Umständen eine Erweiterung des Darmes davor. Eine Fehleinsprossung von Neuronen kann im ganzen Verdauungstrakt vorkommen und dadurch auch Schluckstörungen auslösen.
Herzrhythmusstörungen
Beim CCHS können Sinuspausen (Pausen zwischen zwei Herzschlägen) auftreten.[9] Auch wenn bislang kein direkter Beweis vorliegt, werden Sinuspausen mit Todesfällen in Verbindung gebracht. In mindestens einer wissenschaftlichen Veröffentlichung werden Pausen ab 3 Sekunden als pathologisch angesehen.[9] Bei manchen Patienten wurden im EKG die Zeichen für ein Long-QT-Syndrom gefunden. Klinisch scheint dies nur selten bedeutsam zu werden.
Tumoren
CCHS-Patienten können neuroektodermale Tumoren entwickeln. Dies sind Neubildungen, welche von Nervenzellen oder nervenzellähnlichen Zellen ausgehen. Diese Zellen haben ihren Ursprung in der Neuralleiste des Embryos. Es handelt sich um Neuroblastome und Ganglioneuroblastome.[10]
Temperatur-Regulationsstörungen
Patienten mit einem CCHS können Phasen mit Hyper- oder Hypothermie haben. Zudem treten auch Infektionen ohne Fieber auf.
Behandlung
Etwa 17 % der Undine-Kinder atmen auch während des Wachzustandes nicht ausreichend, sodass sie 24 Stunden am Tag beatmet werden müssen. Viele dieser Kinder sind mit einem Zwerchfellschrittmacher versorgt und dadurch mobil. Atmen Undine-Patienten tagsüber spontan, so bietet sich eine nächtliche Beatmung mit Nasenmaske an. Einige Kinder werden nach Geburt zunächst mit einem Tracheostoma beatmet und im Kindergarten- oder Grundschulalter auf Maske umgestellt.
Heilungsaussichten
Durch die modernen Beatmungs- und Überwachungsmöglichkeiten im häuslichen Bereich können Undine-Kinder heute ein ausgefülltes und produktives Leben führen. So konnte erst kürzlich in einer Studie bei 94 Undine-Kindern in Nordamerika gezeigt werden, dass 77,5 % aller Kinder normale schulische Leistungen erbrachten. Voraussetzung dafür sind eine möglichst frühzeitige Diagnose der Erkrankung und die Versorgung und Schulung der Eltern und der betreuenden Personen mit aktuellen Geräten und Informationen. Da die Diagnose und die Therapiemöglichkeiten dieses Krankheitsbildes erst relativ kurz bekannt sind und zur Verfügung stehen, sind die ältesten Undine-Patienten ca. Anfang zwanzig, vereinzelt jedoch auch Mitte 30. Sie können arbeiten und selbst auch Kinder haben.
Herkunft der Bezeichnung
Die Erstbeschreibung des Syndroms erfolgte 1962 durch Severinghaus und Mitchell.[11] Undine ist der Name einiger in Sagen auftretender Wasserwesen. Davon inspiriert veröffentlichte Friedrich de la Motte Fouqué 1811 eine märchenhafte Erzählung, in der die Nixe Undine einen ihr untreu gewordenen Ritter mit einem Kuss tötet. Aus diesem Stoff entwickelte Jean Giraudoux 1939 sein gleichnamiges Theaterstück, dem er das Thema versagender physiologischer Automatismen beifügte[12]. Eine andere Modifikation erzählt, dass Undine ihren untreuen Mann dazu verflucht habe, dass er im Schlaf nicht mehr atmen könne. Diese Version führte zu der englischen Bezeichnung Ondine's curse, „Undines Fluch“.
Vorkommen in Kunst und Literatur
- Im Film Die schwarze Witwe (1987) wird das Undine-Syndrom als Todesursache eines Mafioso angegeben.
- Am Beginn des Thrillers Der Augensammler von Sebastian Fitzek (2010) wird das Syndrom namentlich erwähnt, erklärt und seine Bezeichnung anhand des Theaterstückes von Giraudoux erläutert; allerdings wird das Thema dort absichtlich falsch auf die germanische Mythologie zurückgeführt.
- Der polnische Kurzfilm Nasza Klątwa (Unser Fluch) erzählt autobiografisch die Geschichte einer jungen Familie, deren Kind mit dem Undine-Syndrom diagnostiziert ist.
Weblinks
- Undine-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Undine Selbsthilfegruppe: Diese Gruppe Betroffener und betroffener Eltern von Kindern mit CCHS hat deutschlandweit und in den Nachbarländern Schweiz und Österreich ca. 83 Mitglieder zwischen Null und 35 Jahren.
- EU CHS Network: Das internationale CHS-Netzwerk ist ein Zusammenschluss von europäischen Ärzten, die über zentrale Hypoventilationssyndrome informieren möchten. Von der EU gefördert stellt es eine Leitlinie zur Diagnose und Therapie des CCHS zusammen, welche noch unveröffentlicht ist. Außerdem ist eine internationale epidemiologische Studie zum Krankheitsbild CCHS Teil der gemeinsamen Arbeit.
- CCHS Network: Das CCHS Network ist die Selbsthilfegruppe in den USA. Die Gruppe ist offen für Mitglieder aus der ganzen Welt.
Einzelnachweise
- Orphanet: Haddad-Syndrom. Abgerufen am 2. November 2018.
- G. G. Haddad u. a.: CONGENITAL FAILURE OF AUTOMATIC CONTROL OF VENTILATION, GASTROINTESTINAL MOTILITY AND HEART RATE. Hrsg.: Medicine. Volume 57, Issue 6, November 1978, S. 517–526.
- S3-Leitlinie Nicht erholsamer Schlaf/Schlafstörungen der Deutschen Gesellschaft für Schlafforschung und Schlafmedizin (DGSM). In: AWMF online (Stand 2009)
- Deutsches Institut für Medizinische Dokumentation und Information: ICD-10-GM Version 2019. Abgerufen am 1. November 2018.
- G. Mayer, Andrea Rodenbeck, Peter Geisler, Hartmut Schulz: Internationale Klassifikation der Schlafstörungen: Übersicht über die Änderungen in der ICSD-3. In: Somnologie - Schlafforschung und Schlafmedizin. Band 19, Nr. 2, 2015, ISSN 1432-9123, S. 116–125, doi:10.1007/s11818-015-0006-8 (PDF download bei Research Gate).
- J. G. Schöber, S. Neumayer, H. Meisner, R. von Kries: Prävalenz und Inzidenz des Undine-Syndroms in Deutschland. In: Monatsschr Kinderheilkd. Band 142, 1994, S. S32.
- PAIRED-LIKE HOMEOBOX 2B; PHOX2B . In: Online Mendelian Inheritance in Man. (englisch)
- Jeanne Amiel, Béatrice Laudier, Tania Attié-Bitach, Ha Trang, Loïc de Pontual: Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. In: Nature Genetics. Band 33, Nr. 4, 17. März 2003, ISSN 1061-4036, S. 459–461, doi:10.1038/ng1130 (nature.com [abgerufen am 1. November 2018]).
- Jerome O. Gronli, Barbara A. Santucci, Sue E. Leurgans, Elizabeth M. Berry-Kravis, Debra E. Weese-Mayer: Congenital central hypoventilation syndrome:PHOX2B genotype determines risk for sudden death. In: Pediatric Pulmonology. Band 43, Nr. 1, 2007, ISSN 8755-6863, S. 77–86, doi:10.1002/ppul.20744 (wiley.com [abgerufen am 20. November 2018]).
- Delphine Trochet, Louise M. O'Brien, David Gozal, Ha Trang, Agneta Nordenskjöld: PHOX2B Genotype Allows for Prediction of Tumor Risk in Congenital Central Hypoventilation Syndrome. In: The American Journal of Human Genetics. Band 76, Nr. 3, März 2005, ISSN 0002-9297, S. 421–426, doi:10.1086/428366, PMID 15657873, PMC 1196394 (freier Volltext) – (elsevier.com [abgerufen am 20. November 2018]).
- J. W. Severinghaus, R. A. Mitchell: Undine's curse – failure of respiratory center automaticity while awake. In: Clin Res. 10, 1962, S. 122.
- R. Nannapaneni, S. Behari. N. V. Todd, A. D. Mendelow: Retracing "Undine's curse". In: Neurosurgery. Band 57, Nr. 2, 2005, S. 354–363; discussion 354–363, doi:10.1227/01.NEU.0000166684.69422.49, PMID 16094167.