Osteopetrose
Die Osteopetrose (auch Osteopetrosis, Marmorknochenkrankheit, Albers-Schönberg-Krankheit, Albers-Schönberg-Syndrom) wird in der Mehrheit der Fälle durch eine erbliche Unterfunktion der knochenabbauenden Zellen (Osteoklasten) verursacht. Durch die ungerichtete Anhäufung von Knochengewebe wird die Mikroarchitektur des Knochens gestört und die mechanische Stabilität gemindert.
| Klassifikation nach ICD-10 | |
|---|---|
| Q78.2 | Marmorknochenkrankheit |
| ICD-10 online (WHO-Version 2019) | |
Da die Osteoklasten im Rahmen der Hämatopoese aus Vorläuferzellen der weißen Blutkörperchen gebildet werden, ist die bisher einzige Therapie eine Knochenmarktransplantation, wonach es zur Bildung funktionsfähiger Osteoklasten und zum Abbau der übermäßigen Sklerosierung kommt, verbunden mit einem beträchtlichen Risiko einer Hypercalcämie.[1]
Klassifikation und Vererbung
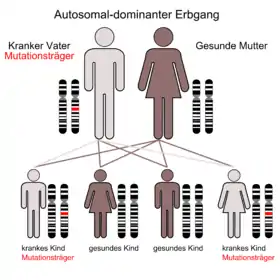
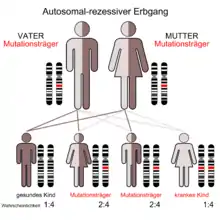
Nach ihrer Vererbung werden die verschiedenen Formen der Osteopetrose in zwei Hauptgruppen klassifiziert:
- autosomal rezessive Formen (infantil maligne Osteopetrose, Osteopetrose mit renal - tubulärer Azidose)
- autosomal dominante Formen (Typ 1 (ADOI), Typ 2 (ADOII, Albers-Schönberg-Krankheit))
Verlauf der autosomal rezessiven Osteopetrose
Die autosomal rezessive Osteopetrose beginnt meist in den ersten beiden Lebensjahren, die Form mit renaler tubulärer Azidose etwas später als die infantil maligne Form, die bereits in den ersten Wochen nach der Geburt zu Symptomen führen kann. Es gibt jedoch auch milder verlaufende rezessive Krankheitsbilder, die mit der dominanten Osteopetrose überlappen können. Unbehandelt ist die Prognose meist schlecht. Die einzige Heilungschance besteht zurzeit in einer Knochenmarktransplantation, da die defekten Osteoklasten aus der hämatopoetischen Linie stammen.
Verlauf der autosomal dominanten Osteopetrose
Für die ADOI ist eine generalisierte Osteosklerose mit besonderer Betonung der Schädelbasis typisch, während für die ADOII, die der ursprünglich von Albers-Schönberg beschriebenen Erkrankung entspricht, die sogenannten „Sandwichwirbel“ im Röntgenbild charakteristisch sind. Beide dominante Formen werden oft während des Wachstumsschubes in der Adoleszenz symptomatisch, wobei die ADOI nicht zu vermehrten Knochenbrüchen, sondern zu einer erhöhten Stabilität führt. Kosmetische Probleme können durch eine Vergrößerung des Unterkiefers entstehen. Die ADOI unterscheidet sich von allen anderen Formen, da sie durch eine Überfunktion der Osteoblasten hervorgerufen wird. Die ADOII führt im Gegensatz hierzu zu häufigen Knochenbrüchen und in Einzelfällen zu Komplikationen ähnlich der malignen Form (v. a. Schädigung der Hirnnerven). Eine kausale Therapie existiert derzeit nicht.
Genetische Ursachen
- Autosomal rezessive Osteopetrose:
Osteoklasten-reiche Formen: TCIRG1, CLCN7, OSTM1, SNX10
Osteoklasten-arme Formen: TNFSF11 (RANKL), TNFRSF11A (RANK)
- Autosomal rezessive Osteopetrose mit renaler tubulärer Azidose: CAII
- Autosomal dominante Osteopetrose Typ 1: LRP5
- Autosomal dominante Osteopetrose Typ 2: CLCN7
Pathogenese
Osteoblasten spielen eine entscheidende Rolle bei der Regulation der Osteoklastenaktivität. Bei Defekt in der CSF-1-Synthese, das von Osteoblasten sezerniert wird, können sich Osteoklasten nicht differenzieren (die Fusionierung der mononukleären Osteoklasten-Progenitorzellen wird unmöglich). Da Osteoklasten nicht vorhanden sind, wird der Knochen nicht abgebaut, was zur Osteopetrose führt.
Auch der Transkriptionsfaktor c-Fos muss in den osteoklastären Progenitorzellen exprimiert werden, damit die weitere Differenzierung erfolgt. Es wurde bei Mäusen das zuständige Gen ausgeschaltet, was zu Osteoklastendefizienz und folglich zu Osteopetrose führte.
Osteoklasten sind im Bereich der sogenannten Haftzone besonders reich an Actinfilamenten und C-Src-Kinase. Die Genausschaltung der C-Src-Kinase führt in Mäusen ebenfalls zur Osteopetrose, da deren Haftung an der Knochenmatrix nicht gegeben ist.
Auch geben Osteoklasten durch Exozytose lysosomale Enzyme ab, unter anderem Tartrat-resistente saure Phosphatase (TRAP) und Cathepsin K, die den Knochenabbau fördern. Der genetisch verursachte Mangel an Cathepsin K führt zur durch Skelettanomalien, Minderwuchs und erhöhter Knochenmasse gekennzeichneten Pyknodysostose, einer wichtigen Differentialdiagnose der Osteopetrose.
Außerdem besitzen Osteoklasten in ihrem Bürstensaum (Ruffled Border) den Chloridkanal ClC-7, der Chlorid in die Lakune zwischen Osteoklast und Knochenmatrix abgibt. Dies ist für den Knochenabbau von zentraler Bedeutung, da Salzsäure in der Lakune gebildet wird. Mutationen von ClC-7 führen zur autosomal rezessiven oder autosomal dominanten Osteopetrose.
Carboanhydrase-II ist ein entscheidendes Enzym in Osteoklasten, das die Bildung von Wasserstoffionen und Hydrogencarbonat aus Wasser und Kohlendioxid ermöglicht. Die Wasserstoffionen werden im Anschluss in die Lakune gepumpt, das Hydrogencarbonat wird durch Chlorid in der glatten Zellseite (Knochenmatrixfern) unter Beteiligung vom Anionenaustauscher 2 (AE2) ausgetauscht. Ein genetischer Defekt in der Carboanhydrase-II führt zu Osteopetrosis.[2]
Klinik
Trotz einer erheblichen Vermehrung der Knochenmasse kommt es zu häufigen Knochenbrüchen, die oft nur schwer verheilen. Als weitere Komplikationen können Leber- und Milzvergrößerung (durch extramedulläre Blutbildung), verringerte Immunabwehr, Krampfanfälle und die Schädigung von Hirnnerven (v. a. Blindheit) auftreten.
Diagnose und Differentialdiagnose
Im Röntgenbild findet sich eine erheblich vermehrte Knochendichte und Verschmälerung des Markraumes. Oft verplumpte Metaphysen der langen Röhrenknochen mit Querstreifen. Verdickung des Schädeldaches und der Schädelbasis.
Abzugrenzen sind
- Pyknodysostose
- Melorheostose
- Sklerostose
- Engelmann-Syndrom (Progressive diaphysäre Dysplasie)
- Pyle-Syndrom[3]
Literatur
- Kornak u. a.: Molekulare Mechanismen der Regulation der Knochendichte durch Osteoklasten. In: Ärzteblatt. Jg. 100, Nr. 19, S. A1258–A1268.
- U. Kornak, S. Mundlos: Genetic disorders of the skeleton: a developmental approach. In: Am. J. Hum. Genet. Band 73, Nr. 3, September 2003, S. 447–474, doi:10.1086/377110, PMID 12900795, PMC 1180673 (freier Volltext).
Einzelnachweise
- Rukshana Shroff, Ortraud Beringer, Kanchan Rao, Lorenz C. Hofbauer, Ansgar Schulz: Denosumab for Post-Transplantation Hyeprcalcemia on Osteopetrosis. In: New England Journal of Medicine. Band 367, Ausgabe 18, 1. November 2012, S. 1766–1767.
- D. Drenkhahn (Hrsg.): Anatomie. Band 1, 16. Auflage. Urban & Fischer, München 2003, S. 133–149.
- F. Hefti: Kinderorthopädie in der Praxis. Springer 1998, ISBN 3-540-61480-X, S. 674.