KBG-Syndrom
Das KBG-Syndrom, auch Hermann-Pallister-Syndrom[1] genannt, ist eine sehr seltene Erbkrankheit.
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.8 | Sonstige näher bezeichnete angeborene Fehlbildungssyndrome, anderenorts nicht klassifiziert |
| ICD-10 online (WHO-Version 2019) | |
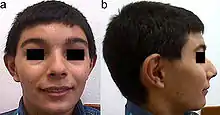
Beschreibung und Symptome
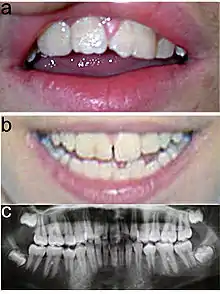
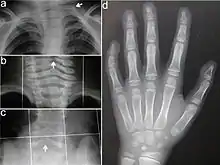
Typisch für das KBG-Syndrom sind faziale Dysmorphien (Gesichtsdysmorphien) und große obere mittlere Schneidezähne. Dazu kommen Anomalien des Skeletts, vor allem costovertebral, das heißt an den Rippenwirbeln, und eine verzögerte körperliche Entwicklung. Letztere führt zu Minderwuchs. Bei der Mehrzahl der Patienten finden sich außerdem EEG-Anomalien (mit oder ohne Krampfanfällen) und ein abnormer Haaransatz. Deutlich seltener sind kutane Syndaktylie, kurzer Hals mit Pterygium, Kryptorchismus („Bauchhoden“), Schwerhörigkeit, Schielen, Gaumendefekt und angeborene Herzfehler.[2]
Häufigkeit
Das KBG-Syndrom ist ausgesprochen selten. Bis 2006 wurden etwa 45 Patienten in 26 Familien weltweit beschrieben. Es gibt offensichtlich mehr männliche Patienten mit dem KBG-Syndrom. Das Geschlechterverhältnis liegt bei ungefähr 2:1. Bei weiblichen Patienten sind die Symptome auch nicht so stark ausgeprägt wie bei männlichen.[2][3]
Genetik
Das KBG-Syndrom wird durch eine heterozygote Mutation im ANKRD11-Gen (Ankyrin Repeat Domain-containing Protein 11), das sich auf Chromosom 16, Genlocus q24.3 befindet, verursacht.[4] Das Genprodukt besteht aus 2663 Aminosäuren, hat eine molare Masse von 298 kDa und beinhaltet unter anderem fünf Ankyrin-Einheiten.[5]
Der Erbgang des KBG-Syndroms ist autosomal-dominant.[6] In einigen Fällen scheint es sich aber auch um spontane Mutationen zu handeln.[3]
Diagnose
Die Diagnosestellung erfolgt im Wesentlichen aufgrund der Symptome, da noch kein Gentest für das KBG-Syndrom verfügbar ist. Dabei orientiert man sich an vier wesentlichen Merkmalen des KBG-Syndroms: faziale Dysmorphie, Makrodentie der oberen mittleren Schneidezähne, skelettale Anomalien (vor allem costovertebral) und verzögerte Entwicklung. Im Alter von sieben bis acht Jahren lässt sich mit der Ausbildung der vorderen Schneidezähne über die Makrodentie die Diagnose sicherer erstellen.[2]
Therapie
Die Behandlung des KBG-Syndroms erfolgt rein symptomatisch. Eventuelle Fehlbildungen können zum Teil chirurgisch korrigiert werden. Zur Betreuung des Patienten gehören regelmäßige EEG-Messungen, eine vollständige kieferorthopädische und skelettale Untersuchung, Hörtests und augenärztliche Kontrollen. Das KBG-Sydrom ist keine lebensbedrohliche Erkrankung.[2] Die Prognose ist daher – auch wenn kaum statistische Daten dazu vorliegen – als günstig einzustufen.
Erstbeschreibung
Das KBG-Syndrom wurde erstmals 1975 von Jürgen Herrmann, Philip David Pallister und John Marius Opitz beschrieben.[7] Von Opitz stammt auch die Namensgebung KBG, die sich aus dem jeweils ersten Buchstaben des Familiennamens seiner drei betroffenen Patienten ergibt.[4]
Weiterführende Literatur
- H. Kumar, N. Prabhu, A. Cameron: KBG syndrome: review of the literature and findings of 5 affected patients. In: Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. Band 108, Nummer 3, September 2009, S. e72–e79, ISSN 1528-395X. doi:10.1016/j.tripleo.2009.04.035. PMID 19716495. (Review).
- K. L. Skjei, M. M. Martin, A. M. Slavotinek: KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. In: American journal of medical genetics. Part A. Band 143, Nummer 3, Februar 2007, S. 292–300, ISSN 1552-4825. doi:10.1002/ajmg.a.31597. PMID 17230487. (Review).
- F. Brancati, M. G. D'Avanzo u. a.: KBG syndrome in a cohort of Italian patients. In: American journal of medical genetics. Part A. Band 131, Nummer 2, Dezember 2004, S. 144–149, ISSN 1552-4825. doi:10.1002/ajmg.a.30292. PMID 15523620.
- S. F. Smithson, E. M. Thompson u. a.: The KBG syndrome. In: Clinical dysmorphology. Band 9, Nummer 2, April 2000, S. 87–91, ISSN 0962-8827. PMID 10826617. (Review).
- I. Morghen, E. Ferri: The KBG syndrome: Case report. In: Cases journal. Band 1, Nummer 1, 2008, S. 186, ISSN 1757-1626. doi:10.1186/1757-1626-1-186. PMID 18822138. PMC 2565666 (freier Volltext).
Einzelnachweise
- R. Witkowski, O. Prokop u. a.: Lexikon der Syndrome und Fehlbildungen. Springer, 2003, ISBN 3-540-44305-3, S. 530. eingeschränkte Vorschau in der Google-Buchsuche
- KBG-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- F. Brancati, A. Sarkozy, B. Dallapiccola: KBG syndrome. In: Orphanet Journal of Rare Diseases. Band 1, 2006, S. 50, ISSN 1750-1172. doi:10.1186/1750-1172-1-50. PMID 17163996. PMC 1764006 (freier Volltext). (Review-Artikel im Open Access).
- KBG-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- KBG-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- M. Tekin, A. Kavaz u. a.: The KBG syndrome: confirmation of autosomal dominant inheritance and further delineation of the phenotype. In: American journal of medical genetics. Part A. Band 130A, Nummer 3, Oktober 2004, S. 284–287, ISSN 1552-4825. doi:10.1002/ajmg.a.30291. PMID 15378538. (Review).
- J. Herrmann, P. D. Pallister u. a.: The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. In: Birth defects original article series. Band 11, Nummer 5, 1975, S. 7–18, ISSN 0547-6844. PMID 1218237.