Gallengangatresie
Die Gallengangatresie (englisch: biliary atresia, BA) ist eine seltene Erkrankung mit Verschluss (Atresie) der Gallenwege, die ausschließlich im Neugeborenenalter (Neonatalperiode) auftritt. Die Ursache ist noch ungeklärt. In den entwickelten Ländern stellt die Gallengangatresie die häufigste Ursache für die Notwendigkeit einer Lebertransplantation im Säuglingsalter dar.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| Q44.2 | Atresie der Gallengänge |
| ICD-10 online (WHO-Version 2019) | |
Vorkommen
Die Prävalenz bei Geburt variiert weltweit zwischen 1:20.000 und 1:3.100 Lebendgeborene, wobei eine Häufung der Erkrankung in Asien und im Pazifikraum beobachtet wurde. In Westeuropa erkrankt etwa ein Kind von 18.000 innerhalb der Neonatalperiode.[2] Mädchen sind etwas häufiger betroffen als Jungen. Etwa ein Zehntel der Fälle ist mit zusätzlichen angeborenen Fehlbildungen (z. B. Herzfehler, Polysplenie) vergesellschaftet und wird als syndromale Form zusammengefasst. Das alleinige Auftreten des Verschlusses der Gallenwege wird als nicht-syndromale Form bezeichnet. International hat sich durchgesetzt, die Unterteilung zwischen extra- und intrahepatischer Form nicht mehr zu treffen, da die Erkrankung die gesamte Leber und alle Gallengänge betrifft.
Ursache
Die Ätiologie (Ursache) der Gallengangatresie ist weitgehend unbekannt. Verschiedene Hinweise – beispielsweise Veränderungen der Ultraschallstruktur der Leber schon im Mutterleib – legen nahe, dass die Verengung der Gallenwege schon früh in der Schwangerschaft beginnt. Umfangreiche Untersuchungen über den Einfluss von Virusinfektionen haben einen Zusammenhang mit Cytomegalie-, Respiratory-Syncitial-, Epstein-Barr- und humanen Papilloma-Viren unterstellt. Dahingegen konnte keine Verbindung zu den Hepatitis-Viren A, B oder C gefunden werden. Auch genetische Einflüsse scheinen eine Rolle zu spielen: einzelne Studien berichten über familiäre und ethnische (im Pazifikraum oder Teilen der USA) Häufungen, ebenso wurden bestimmte HLA-Typen (HLA-B12; Haplotyp A9-B5; Haplotyp A28-B35) gehäuft bei betroffenen Kindern gefunden.[2] Bei der feingeweblichen (histopathologischen) Untersuchung unter dem Mikroskop findet man eine entzündliche Schädigung der Gallengänge mit Vermehrung des Bindegewebes (Sklerose) und Verengung bis hin zur Verlegung der Gallenwege.
Symptome
Nach der Geburt entwickeln die Kinder eine verlängerte Gelbsucht (Ikterus), die im Gegensatz zur normalen Neugeborenengelbsucht überwiegend durch wasserlösliches „direktes“ Bilirubin verursacht wird. Sie setzen entfärbten Kot (sogenannter acholischer Stuhl) ab und der Urin färbt sich braun. Als drittes Leitsymptom kann eine Vergrößerung der Leber (Hepatomegalie) beobachtet werden. Es findet sich sehr häufig eine Assoziation mit fazialen Dysmorphien, Augenfehlbildungen, Herzvitien und Skelettfehlbildungen. Der Allgemeinzustand der Kinder ist dabei zunächst gut. Nicht einmal das Gedeihen ist in den ersten Monaten beeinträchtigt. Erst später setzt ein Gewichtsverlust und eine zunehmende Übererregbarkeit ein. Als Zeichen eines Druckanstiegs in der Pfortader der Leber (Portale Hypertension) kommen schließlich eine Milzvergrößerung und Wasseransammlungen im Bauchraum (Aszites) hinzu. Weil mit dem gestörten Gallefluss auch zu wenig Gallensäuren in den Darm gelangen, ist die Fettverdauung und damit auch die Aufnahme fettlöslicher Vitamine, insbesondere des Vitamin K gestört, was zu einer Blutungsneigung führen kann.
Diagnose
Bei jedem Kind mit einer Neugeborenengelbsucht, die länger als zwei Wochen andauert, muss wegen der besonderen prognostischen Bedeutung einer frühen Diagnosestellung eine Gallengangatresie aktiv ausgeschlossen werden. Dazu gehört als erster Schritt eine laborchemische Differenzierung des Bilirubins in die wasserlösliche, konjugierte (direkte) und die wasserunlösliche, unkonjugierte (indirekte) Form. Bei einer Ultraschalluntersuchung nach einer vier- bis zwölfstündigen Fastenperiode erhärtet eine nicht darstellbare oder geschrumpfte Gallenblase, eine vermehrte Echogenität des Leberhilus oder eine Zyste im Leberhilus den Verdacht. Bei normaler Gallenblase im Ultraschall und fortbestehendem Verdacht muss der anatomische Aufbau und die Durchgängigkeit der Gallenwege mit einer Röntgen-Kontrastmittel-Darstellung, einer Cholangiografie untersucht werden. Letzte diagnostische Unsicherheiten können gegebenenfalls durch eine Leberbiopsie geklärt werden.
Behandlung
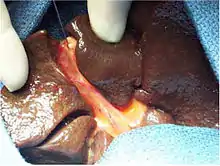
Unbehandelt führt die Erkrankung zu einem allmählichen bindegewebigen Umbau der Leber (Leberzirrhose) und dem Tod innerhalb der ersten Lebensjahre. Um den Gallefluss behelfsweise wiederherzustellen, wird bei den betroffenen Kindern zunächst eine Operation nach Kasai, eine Hepatoporto-Enterostomie, vorgenommen. Dabei werden die veränderten Gallenwege einschließlich des Bindegewebes zwischen dem rechten und linken Pfortaderast in der Leberpforte entfernt. Anschließend wird eine Darmschlinge auf die offene Leberpforte aufgenäht, so dass die Gallenflüssigkeit sozusagen aus der Leberpforte direkt in den Darm ablaufen kann. Die zusätzliche medikamentöse Behandlung entweder mit entzündungshemmenden Mitteln, die den zunehmenden Umbau des Lebergewebes verlangsamen sollen, oder mit Substanzen, die den Gallefluss verbessern können, werden verschiedentlich empfohlen, sind aber aufgrund eines fehlenden Nachweises eines Langzeitnutzens umstritten.
Erholt sich der Gallefluss durch die Kasai-Operation, zeigt sich dies in einem Rückgang der Gelbsucht und zunehmender Braunfärbung des Kotes. Doch selbst wenn diese günstige Situation eintritt, entwickeln zwei Drittel der Patienten eine durch den Gallestau verursachte Leberzirrhose. Diese oder ein primäres Versagen der Kasai-Operation macht eine Lebertransplantation erforderlich. Das Organ wird zumeist im zweiten Lebensjahr verpflanzt, was aber auch schon im Alter von sechs Monaten erforderlich werden kann.[2] Durch die Entwicklung neuer Transplantationsverfahren (Leber-Splitting, Lebend-Spende) ist die Verfügbarkeit dieser Behandlungsmethode in jüngster Zeit angestiegen.
Prognose
Ein Überleben mit der eigenen Leber bis ins Erwachsenenalter wird nur bei etwa einem Zehntel der Patienten beobachtet. Dennoch können heutzutage insgesamt etwa 90 % aller Betroffenen auf ein Überleben mit weitestgehend normaler Lebensqualität hoffen.[2] Gallengangatresien, die mit zusätzlichen Fehlbildungen der Milz einhergehen, haben dabei grundsätzlich eine schlechtere Prognose. Genauso sinkt die Aussicht auf eine erfolgreiche Behandlung, je weiter die Veränderungen der Gallenwege in die Leber hineinreichen. Die Erfolgsaussicht für die Kasai-Operation sinkt wiederum mit zunehmendem Alter der Kinder, weshalb eine frühe Diagnosestellung von besonderer Bedeutung ist. Darüber hinaus verbessert sich die Gesamtprognose mit der Verfügbarkeit einer Lebertransplantation. Im Hamburger Transplantationszentrum betrug die Überlebensquote von Kindern, die im Säuglingsalter eine Lebertransplantation erhalten hatten, nach zwei Jahren 87 %.[1]
Einzelnachweise
- E. F. Grabhorn u. a.: Lebertransplantation im Säuglingsalter. In: Monatsschrift Kinderheilkunde. 2007, 155, S. 381–389. doi:10.1007/s00112-007-1485-x.
- C. Chardot: Biliary atresia. Review In: Orphanet Journal of Rare Diseases. 2006; 1, S. 28. PMC 1560371 (freier Volltext).