Fallot-Tetralogie
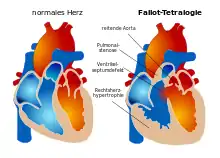
Die Fallot-Tetralogie (auch Fallot’sche Tetralogie) ist eine angeborene Herzfehlbildung, die etwa 6–7 % der angeborenen Herzfehler ausmacht. Sie besteht aus vier Komponenten (daher Tetralogie): einer Pulmonalstenose, einem Ventrikelseptumdefekt (Defekt in der Herzscheidewand), einer über der Herzscheidewand reitenden Aorta sowie einer nachfolgenden Rechtsherzhypertrophie. Die auch im Deutschen bei Medizinern gebräuchliche Abkürzung TOF leitet sich aus dem Englischen Tetralogy of Fallot ab. In etwa einem Drittel der Fälle treten neben den Herzveränderungen weitere Fehlbildungen auf, vor allem am Verdauungstrakt sowie Spalten.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| Q21.3 | Fallot-Tetralogie Ventrikelseptumdefekt mit Pulmonalstenose oder -atresie, Dextroposition der Aorta und Hypertrophie des rechten Ventrikels |
| ICD-10 online (WHO-Version 2019) | |
Geschichte
Die Bezeichnung dieses Herzfehlers geht auf den französischen Pathologen Étienne-Louis Arthur Fallot als Beschreiber (1888) zurück. Die erste Beschreibung der Fallot-Tetralogie stammt aus dem Jahre 1672 von Stensen,[2] eine weitere von Eduard Sandifort (1777). Auch der Pathologe Carl von Rokitansky beschrieb (1876) diese Missbildung.[3] Der englische Chirurg Thomas Bevill Peacock (1812–1882) beschrieb sie 1858[4] in der ersten ausführlichen Abhandlung über angeborene Herzfehlbildungen.[5]
1945 wurde durch Blalock und Taussig erstmals eine Fallot-Tetralogie durch eine angelegte Verbindung (Shunt) zwischen Arteria subclavia und Pulmonalarterie chirurgisch korrigiert. 1946 stellten Potts und Mitarbeiter eine Anastomose zwischen Aorta und linker Arteria pulmonalis her, 1962 Waterson zwischen Aorta und rechter Arteria pulmonalis.[2]
Entstehung
Eigentlich liegen bei dieser Fehlbildung keine vier separaten Defekte vor, sondern ein einziger, nämlich eine Verlagerung der infundibulären Scheidewand der rechten Herzkammer nach vorn.[6]
Am Ende der fünften Entwicklungswoche entspringt die Ausstrombahn des Herzens noch aus beiden Herzhälften. Durch Ausbildung von Endokardwülsten im Conus cordis entsteht das Septum aorticopulmonale, das den Conus cordis in rechte und linke Ausstrombahn teilt (Aorta und Truncus pulmonalis). Verlagert sich jedoch das Septum aorticopulmonale nach vorne, verengt sich das Kammerausflussgebiet (Pulmonalstenose), das Septum ist allerdings noch geöffnet (Ventrikelseptumdefekt). Die Aorta entspringt nun aus beiden Herzkammern (reitende Aorta). Durch die Pulmonalstenose kommt es zu Bluthochdruck (arterielle Hypertonie) in der rechten Herzkammer. Damit noch genügend Blut aus dem Herzen gepumpt werden kann, vergrößert sich die Kammerwand (hypertrophierter rechter Ventrikel).
In 10 % der Fälle ist die Fallot-Tetralogie mit einer Chromosomenaberration assoziiert, am häufigsten mit einem Edwards-Syndrom oder einem Down-Syndrom.[1]
Die Fehlbildungen
Folgende Fehlbildungen bilden das Krankheitsbild:
Pulmonalstenose
Die Pulmonalstenose als Verengung des Ausgangstraktes der rechten Herzkammer zur Lunge. Die einzelnen Anteile dieses Ausflusstraktes (herznaher Anteil, Klappenring mit halbmondförmigen Klappen (semilunares) und lungennaher Anteil) können unterschiedlich stark verengt sein. Die eigentliche Pulmonalklappe ist häufig missgebildet und weist in über 60 % der Fälle nur zwei Halbmondklappen auf. Deshalb kann der Schweregrad der Klappenverengung sehr stark variieren. Das Pulmonalgefäßsystem ist - entsprechend den Verengungen im gesamten Ausflusstrakt - in der Regel unterentwickelt.
Ventrikelseptumdefekt
Der Ventrikelseptumdefekt ist ein Loch in der Scheidewand zwischen den Herzkammern, der bei der Fallot-Tetralogie nicht ein Mangel an Substanz der Kammerscheidewand ist, sondern durch die Fehlbildung des Herzens entstand. Er liegt meist direkt unterhalb des rechtsventrikulären Ausflusstraktes (Pulmonalklappe) und in der Nähe der Aortenklappe (Auslassklappe für den Körperkreislauf) und der Trikuspidalklappe (Klappe zwischen rechter Hauptkammer und rechter Vorkammer).
Überreiten der Aorta
Durch eine mangelhafte Rotation des Pulmonalgefäßes in der Herzentwicklung bleibt die Aortenwurzel in ihrer Beziehung zur rechten Herzkammer positioniert, so dass ein „Überreiten der Aorta“ über dem Ventrikelseptumdefekt resultiert.
Rechtsherzhypertrophie
Die Rechtsherzhypertrophie, ein die Herztätigkeit behindernder Zuwachs der Muskelmasse des rechten Herzens aufgrund der chronischen Überbelastung, kann anfangs gering sein, jedoch im Laufe der Zeit zunehmen.
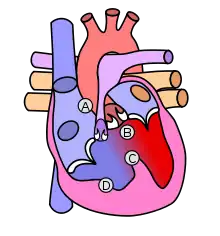 A: Pulmonalstenose, B: überreitende Aorta, C: Ventrikelseptumdefekt, D: Rechtsherzhypertrophie |
 Gesundes Herz |
Folgen für das Herz-Kreislauf-System
Das über den rechten Herzvorhof aus dem Körper zurückkommende sauerstoffarme (venöse) Blut gelangt auf Grund der Verengung des pulmonalen Ausflusstraktes nur teilweise in die Lunge, um dort mit Sauerstoff angereichert zu werden. Ein entsprechend größerer Anteil weicht über den großen Ventrikelseptumdefekt aus und fließt unter Umgehung des Lungenkreislaufes direkt über die Aorta (große Körperschlagader) wieder in den Körper. Ein solcher Übertritt von Blut vom rechten Herzen direkt in den Körperkreislauf wird Rechts-Links-Shunt genannt.
Symptome
Durch die Mischung von sauerstoffarmem Blut über den Shunt mit dem aus der Lunge zurückkehrenden sauerstoffreichen Blut entsteht eine Zyanose. Das ist an einer Blaufärbung von Haut und Schleimhäuten, hauptsächlich an Mund und Händen/Füßen, sichtbar, durch den Sauerstoffmangel im Körperkreislauf. Sie kann je nach Menge des übertretenden Blutes mehr oder weniger ausgeprägt sein und wird möglicherweise nur nach Belastung (bei kleinen Kindern Trinken oder Schreien) sichtbar. Ist die Zyanose gering, spricht man auch von einem „Pink Fallot“, bei dem samtartige, tiefrote Schleimhäute zu sehen sein können. Eine im Verlauf der ersten Lebensmonate (nach Schließen des Ductus arteriosus) auftretende schwere Zyanose lässt bereits bei der Betrachtung des Patienten eine Missbildung des Herzens vermuten. Bei der Palpation ist ein systolisches Schwirren über der Herzbasis zu ertasten.
Auf den Mangel an Sauerstoff im Blut reagiert der Körper mit einer Vermehrung der Erythrozyten (rote Blutkörperchen). In gewissen Grenzen kann damit das Sauerstoffdefizit ausgeglichen werden. Aber die Fließeigenschaft des Blutes wird ebenfalls verändert, und es können sich kleine Gerinnsel bilden, die eine Embolie auslösen können. Auch das Wachstum der kleinen Gefäße an Fingern und Zehen nimmt zu. Es können sich so genannte Trommelschlägelfinger und -zehen sowie Uhrglasnägel entwickeln. Wegen der verschlechterten Fließeigenschaft des Blutes sollte den Kindern immer ausreichend zu trinken angeboten werden. Die bestehende Rechtsherzhypertrophie (Vergrößerung der rechten Herzkammer) ist bei der Perkussion erkennbar (Holzschuhform des Herzens).
Hypoxämische Anfälle
Eine Besonderheit der Fallot-Tetralogie sind hypoxämische Anfälle. Sie treten nur bei einem kleinen Teil der Kinder auf, werden jedoch kaum bei Kindern mit anderen zyanotischen Herzfehlern beobachtet. Bei einem solchen Anfall werden die Kinder unruhig und ängstlich, die Zyanose nimmt zu, die Herzfrequenz steigt auf Werte zwischen 140 und 160 Schläge/Minute. Die Kinder können grau-blass-blau erscheinen und das Bewusstsein ist getrübt, bis hin zur Bewusstlosigkeit. Die Anfälle entwickeln sich meist ohne erkennbare äußere Anlässe und die Ursache ist nicht immer eindeutig erkennbar.
Ein hypoxämischer Anfall muss notfallmäßig behandelt werden. Sein Auftreten erfordert meist eine baldige Operation. Unter häuslichen Bedingungen ist frische Luft und eine Beruhigung als Erste Hilfe geeignet. In der Klinik wird ein Anfall mit Sauerstoffgaben, medikamentöser Sedierung (etwa durch Opioide) und der Gabe von Betablockern (etwa Propranolol; kann prophylaktisch wirken) behandelt.
Diagnostik
Über der Herzbasis ist bei der Auskultation eine lautes (bei der Phonokardiographie auch spindelförmiges), systolisches Geräusch, am lautesten links vom Brustbein zu hören. Zur Diagnostik stehen heute meist die Echokardiografie und seltener auch die Herzkatheteruntersuchung zur Verfügung.[7] Das verwandte Krankheitsbild eines Truncus arteriosus communis kann bei einer Spaltung des 2. Herztons ausgeschlossen werden.[8]
Therapie
Die Therapie besteht in einer chirurgischen Korrektur. Es wird angestrebt, eine normale Anatomie zu schaffen. Die Pulmonalstenose wird erweitert und der Ventrikelseptumdefekt durch einen Patch (Flicken) – in der Regel aus Perikard oder Goretex – so verschlossen, dass die Aorta ausschließlich sauerstoffreiches Blut aus dem linken Ventrikel (Herzkammer) erhält. Angestrebt wird eine Operation in einem Schritt im ersten Lebensjahr. Es hängt aber von den Einzelfaktoren bei dem Patienten ab, ob dieses Vorgehen möglich ist. Um ein höheres Alter für die Korrektur zu erreichen, wurde früher zunächst oft eine Blalock-Taussig-Anastomose angelegt. Bei besonderen Konstellationen kann es auch heute noch angezeigt sein, eine Palliativoperation durch Anlage eines aorto-pulmonalen Shunts der Hauptoperation voranzustellen. Am häufigsten wird ein modifizierter Blalock-Taussig-Shunt in Form einer Goretex-Prothese zwischen Truncus brachiocephalicus und rechter Pulmonalarterie oder ein zentraler Shunt zwischen Aorta und Pulmonalarterie angelegt. Diese Operation gilt als leichterer Eingriff und wird in der Regel ohne Einsatz der Herz-Lungen-Maschine durchgeführt. Dadurch wird die Lungendurchblutung vermehrt und damit die Sauerstoffversorgung verbessert. Bei der späteren endgültigen Operation wird der Shunt wieder verschlossen. In einigen Fällen kann bei einem Säugling im Rahmen einer Herzkatheterintervention in Form des Aufdehnens der Pulmonalstenose mit einem Ballon die Operation um einige Wochen oder Monate aufgeschoben werden.
Behandlungsergebnisse und Kontrolluntersuchungen
In der Regel kommt es nach der Operation zu einem komplikationslosen Verlauf und die Herzkammern können sich normal entwickeln. Regelmäßige Kontrollen (alle sechs bis zwölf Monate, lebenslang) durch den Kinderkardiologen sollten unbedingt durchgeführt werden, um eine möglicherweise verbliebene geringe Pulmonalstenose oder Undichtigkeit der Klappe zu beobachten. Diese können aktuell in vielen Fällen katheterinterventionell durch Einbringen einer neuen biologischen Herzklappe in Position der Pulmonalklappe ohne erneute Operation behandelt werden.[9] Auch auf auftretende Herzrhythmusstörungen wird bei diesen Kontrolluntersuchungen geachtet. Eine Endokarditisprophylaxe muss lebenslang beachtet werden.
Es ist zu erwarten, dass sich die heute operierten Kinder normal entwickeln und im Erwachsenenalter eine normale körperliche Belastbarkeit und gute Lebensqualität erreichen. Jedoch kann der Verlauf, wie bei allen komplexen angeborenen Herzfehlern, unterschiedlich sein. Früher operierte Patienten, bei denen die Korrekturoperation aus technischen Gründen (fehlende frühe Diagnose- und Operationsmöglichkeiten) erst im Kindes- oder Jugendlichenalter vorgenommen wurde, sind möglicherweise nicht mit den heutzutage operierten Kindern zu vergleichen.
Daten und Fakten
- Spontanverlauf ist abhängig vom Ausmaß der Lungendurchblutung, Menschen mit dieser Krankheit (ohne Operation) besitzen eine mittlere Lebenserwartung von 12 Jahren, 95 % sterben vor dem 40. Lebensjahr.[10]
- Geschlechtsverhältnis: männlich: weiblich= 1,4 : 1[10]
- Etwa 15 % der Patienten weisen eine Mikrodeletion an Chromosom 22q11 auf.[10]
Siehe auch
Literatur
- S2k-Leitlinie Fallotsche Tetralogie der Deutschen Gesellschaft für Pädiatrische Kardiologie (DGPK). In: AWMF online (Stand 2013–2018)
- F. Bailliard, R. H. Anderson: Tetralogy of Fallot. In: Orphanet Journal of Rare Diseases. Band 4, 2009, S. 2, ISSN 1750-1172. doi:10.1186/1750-1172-4-2. PMID 19144126. PMC 2651859 (freier Volltext). (Review).
- E. A. Shinebourne, S. V. Babu-Narayan, J. S. Carvalho: Tetralogy of Fallot: from fetus to adult. In: Heart. Band 92, Nummer 9, September 2006, S. 1353–1359, ISSN 1468-201X. doi:10.1136/hrt.2005.061143. PMID 16908723. PMC 1861206 (freier Volltext). (Review).
- F. A. Pigula et al.: Repair of tetralogy of Fallot in neonates and young infants. In: Circulation. Band 100, Nummer 19 Suppl, November 1999, S. II157–II161, ISSN 0009-7322. PMID 10567296.
- Klaus Holldack, Klaus Gahl: Auskultation und Perkussion. Inspektion und Palpation. Thieme, Stuttgart 1955; 10., neubearbeitete Auflage ebenda 1986, ISBN 3-13-352410-0, S. 187–190 und 196 f.
Weblinks
- Fallot-Tetralogie. In: Orphanet (Datenbank für seltene Krankheiten).
- Animierte Herzfehlerbeschreibung einer Fallotschen Tetralogie. herzklick.de
Einzelnachweise
- Christof Sohn at al.: Ultraschall in Gynäkologie und Geburtshilfe. Georg Thieme, Stuttgart 2003, ISBN 978-3-13-101972-1, S. 195.
- Christof Schmid: Leitfaden Kinderherzchirurgie. Springer, Berlin 2013, ISBN 978-3-662-12258-7, S. 47.
- Klaus Holldack, Klaus Gahl: Auskultation und Perkussion. Inspektion und Palpation. Thieme, Stuttgart 1955; 10., neubearbeitete Auflage ebenda 1986, ISBN 3-13-352410-0, S. 100 und 187.
- T. B. Peacock: On malformations […] of the human heart. London 1858.
- Barbara I. Tshisuaka: Peacock, Thomas Bevill. In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2005, ISBN 3-11-015714-4, S. 1119.
- Christof Schmid: Leitfaden Kinderherzchirurgie. 2013, S. 48.
- Fallotsche Tetralogie S2k. Deutsche Gesellschaft für Pädiatrische Kardiologie (DGPK), AWMF, 2013–2018 (archivierte Version abgerufen am 16. Februar 2020)
- Klaus Holldack, Klaus Gahl: Auskultation und Perkussion. Inspektion und Palpation. Thieme, Stuttgart 1955; 10., neubearbeitete Auflage ebenda 1986, ISBN 3-13-352410-0, S. 189 f. und 196 f.
- Luca Oechslin, Roberto Corti, Matthias Greutmann, Oliver Kretschmar, Oliver Gaemperli: Percutaneous pulmonary valve implantation in grown-up congenital heart disease patients: Insights from the Zurich experience. In: Journal of Interventional Cardiology. Band 31, Nr. 2, 2018, ISSN 1540-8183, S. 251–260, doi:10.1111/joic.12477 (wiley.com [abgerufen am 6. Februar 2019]).
- Gerd Herold: Innere Medizin. Köln 2015, ISBN 978-3-9814660-8-9, S. 193.