Diwasserstoff-Kation
Das Wasserstoff-Molekülion, Diwasserstoff-Kation, oder H2+, ist das einfachste Molekülion. Es besteht aus zwei positiv geladenen Protonen und einem negativ geladenen Elektron und kann durch Ionisierung des neutralen Wasserstoff-Moleküls gebildet werden.
Es ist von großem historischem und theoretischem Interesse, da es nur ein Elektron enthält und deshalb keine Elektron-Elektron-Wechselwirkungen auftreten. Daher lässt sich die elektronische Schrödinger-Gleichung für dieses System bei festgehaltenem Kernabstand (sog. Born-Oppenheimer-Näherung) in geschlossener Weise analytisch lösen. Die analytischen Lösungen für die Energie-Eigenwerte[1] stellen eine Verallgemeinerung der lambertschen W-Funktion dar.
Wegen seiner Bedeutung als einfachstes molekulares System wird das Wasserstoff-Molekülion in den meisten Lehrbüchern der Quantenchemie als Beispiel behandelt. Die erste erfolgreiche quantenmechanische Behandlung des H2+ wurde vom dänischen Physiker Øyvind Burrau im Jahr 1927 veröffentlicht,[2][3] gerade ein Jahr nach der Veröffentlichung der grundlegenden Arbeit zur Wellenmechanik durch Erwin Schrödinger. Frühere Versuche waren im Jahr 1922 durch Karel Niessen[4] und Wolfgang Pauli,[5] und im Jahr 1925 durch Harold Urey[6] veröffentlicht worden. Mit einem Übersichtsartikel aus dem Jahr 1928 machte Linus Pauling sowohl die Arbeit von Burrau als auch die von Walter Heitler und Fritz London über das Wasserstoffmolekül einem größeren Leserkreis bekannt.[7]
Die chemische Bindung in H2+ kann als kovalente Ein-Elektron-Bindung beschrieben werden, die eine formale Bindungsordnung von 1/2 hat.[8]
Das Wasserstoff-Molekülion wird gewöhnlich auch in Molekülwolken im Weltall gebildet und ist von großer Bedeutung für die Chemie im interstellaren Medium.
Quantenmechanische Behandlung, Symmetrien und Asymptotik
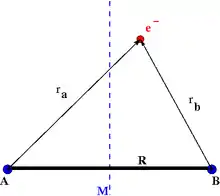
Die einfachste elektronische Schrödinger-Gleichung für das Wasserstoff-Molekülion berücksichtigt neben dem einen Elektron die beiden Kerne, gekennzeichnet mit A und B, an festen Positionen im Raum. Sie kann geschrieben werden als

wobei die Elektron-Kern-Coulomb-Potentialfunktion


ist, und ist die (elektronische) Energie eines gegebenen quantenmechanischen Zustands (Eigenzustands), mit der elektronischen Zustandsfunktion die von den Ortskoordinaten des Elektrons abhängt. Ein additiver Term , der für vorgegebenen Kern-Kern-Abstand eine Konstante ist, wurde in der Potentialfunktion fortgelassen, da er den Eigenwert nur verschiebt. Die Abstände zwischen dem Elektron und den Kernen seien mit und bezeichnet. In atomaren Einheiten wird die Schrödinger-Gleichung zu








Der Mittelpunkt zwischen den Positionen der Kerne kann als Ursprung der Koordinaten gewählt werden. Aus allgemeinen Symmetrieprinzipien folgt, dass die Zustandsfunktionen nach ihrem Symmetrieverhalten bezüglich Rauminversion charakterisiert werden können. Es gibt Zustandsfunktionen

- ,

die "symmetrisch" bezüglich Rauminversion sind, und Zustandsfunktionen
- ,

die unter dieser Symmetrieoperation anti-symmetrisch sind:

Wir merken an, dass die Permutation (der Austausch) der Kerne die gleiche Wirkung auf die elektronischen Zustandsfunktionen hat. Für ein Mehrelektronensystem muss, zusätzlich zu diesen gerade benannten Symmetrien, auch das richtige Symmetrieverhalten der Zustandsfunktion bezüglich Permutationen der Elektronen (Paulisches Ausschließungsprinzip) gewährleistet sein. Die Schrödinger-Gleichungen für die symmetrieangepassten Zustandsfunktionen sind nun


Der Grundzustand (der energetisch niedrigste diskrete Zustand) des ist der Zustand[9], die zugehörige Zustandsfunktion wird üblicherweise mit gekennzeichnet. Die Zustandsfunktion des ersten angeregten Zustands, , wird mit gekennzeichnet. Die hier auftretenden Suffixe g und u (von gerade und ungerade) kennzeichnen gerade das Symmetrieverhalten unter Rauminversion. Ihre Verwendung ist Standard für die Kennzeichnung elektronischer Zustände von zweiatomigen Molekülen, während für Zustände von Atomen die Kennzeichnungen e und o (von Englisch „even“ und „odd“) verwendet werden.







Für große Kern-Kern-Abstände haben die (totalen) Energie-Eigenwerte für diese beiden niedrigsten Zustände dieselbe asymptotische Entwicklung in reziproken Potenzen des Kern-Kern-Abstandes R:[10]


Die tatsächliche Differenz zwischen diesen beiden Energien wird Austauschenergieaufspaltung genannt und ist gegeben durch:[11]

Dieser Ausdruck verschwindet exponentiell mit Zunahme des Kern-Kern-Abstandes. Der führende Term wurde erst mit der Holstein-Herring-Methode richtig erhalten. In ganz ähnlicher Weise wurden asymptotische Entwicklungen in Potenzen von bis zu hoher Ordnung von Čížek et al. für die niedrigsten zehn diskreten Zustände des Wasserstoff-Molekülions erhalten (für den Fall festgehaltener Kerne). Für beliebige zweiatomige oder mehratomige molekulare Systeme lässt sich die Austauschenergieaufspaltung bei großem Kern-Kern-Abstand nur sehr schwer berechnen. Für die Behandlung langreichweitiger Wechselwirkungen, einschließlich Studien mit Bezug auf Magnetismus und Ladungsaustauscheffekte, ist ihre Kenntnis aber notwendig. Die genannten Effekte sind insbesondere von Bedeutung für das physikalische Verständnis von Sternen und von Atmosphären (terrestrisch und extraterrestrisch).

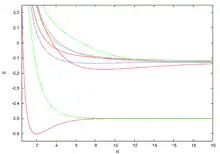
Die Energien für die niedrigsten diskreten Zustände sind in der obigen Abbildung gezeigt. Die Werte können mit jeder gewünschten Genauigkeit unter Verwendung eines Computeralgebraprogramms aus der „verallgemeinerten“ lambertschen W-Funktion erhalten werden (siehe Gl. dort und die Referenz auf die Arbeit von Scott, Aubert-Frécon, und Grotendorst) doch sie wurden zunächst numerisch erhalten, in doppelter Genauigkeit, mit Hilfe des genauesten verfügbaren Computerprogrammes genannt ODKIL.[12] Die roten durchgezogenen Linien sind Zustände. Die grünen gestrichelten Linien sind Zustände. Die blaue gestrichelte Linie ist ein Zustand, und die rosa gepunktete Linie ist ein Zustand. Obwohl die mit Hilfe der „verallgemeinerten“ lambertschen W-Funktion erhaltenen Eigenwertlösungen diese asymptotischen Entwicklungen ersetzen, sind sie in der Praxis besonders in der Umgebung des Gleichgewichtsabstands sehr brauchbar. Solche Lösungen sind möglich, weil die partielle Differentialgleichung, die die Schrödinger-Gleichung darstellt, unter Verwendung von prolaten sphäroidalen Koordinaten in zwei gekoppelte gewöhnliche Differentialgleichungen separierbar ist.





Bildung
Das Wasserstoff-Molekülion wird in der Natur durch die Wirkung kosmischer Strahlung auf Wasserstoffmoleküle gebildet. Ein Elektron wird dabei herausgeschlagen und lässt das Kation zurück.[13]

Die Teilchen der kosmischen Strahlung besitzen genügend Energie, um viele Moleküle zu ionisieren, bevor sie selbst abgestoppt werden.
Der maximale Wirkungsquerschnitt beträgt für sehr schnelle Protonen (70 keV) 2,5 × 10−16 cm2.
In der Natur reagiert das Ion weiter mit anderen Wasserstoffmolekülen:

Eigenschaften
Die Ionisierungsenergie des Wasserstoffmoleküls beträgt , die Dissoziationsenergie .


Einzelnachweise
- T. C. Scott, M. Aubert-Frécon, J. Grotendorst: New Approach for the Electronic Energies of the Hydrogen Molecular Ion. Im: Chem. Phys. 324, 2006, S. 323–338, doi:10.1016/j.chemphys.2005.10.031, arxiv:physics/0607081.
- Ø. Burrau: Berechnung des Energiewertes des Wasserstoffmolekel-Ions (H2+) im Normalzustand. In: Danske Vidensk. Selskab. Math.-fys. Meddel.. M 7:14, 1927, S. 1–18.
- Øjvind Burrau: The calculation of the Energy value of Hydrogen molecule ions (H2+) in their normal position. In: Naturwissenschaften. 15, Nr. 1, 1927, S. 16–17. doi:10.1007/BF01504875.
- Karel F. Niessen Zur Quantentheorie des Wasserstoffmolekülions, Dissertation, Universität Utrecht, Utrecht: I. van Druten (1922), zitiert in J. Mehra, Volume 5, Part 2, 2001, S. 932.
- W. Pauli: Über das Modell des Wasserstoffmolekülions. In: Ann. d. Phys.. 373, Nr. 11, 1922, S. 177–240. doi:10.1002/andp.19223731101. erweiterte Dissertation; eingegangen 4 März 1922, veröffentlicht im Heft Nr. 11 vom 3. August 1922.
- Urey HC: The Structure of the Hydrogen Molecule Ion. In: Proc. Natl. Acad. Sci. U.S.A.. 11, Nr. 10, Oktober 1925, S. 618–21. doi:10.1073/pnas.11.10.618. PMID 16587051. PMC 1086173 (freier Volltext).
- L. Pauling: The Application of the Quantum Mechanics to the Structure of the Hydrogen Molecule and Hydrogen Molecule-Ion and to Related Problems. In: Chemical Reviews. 5, 1928, S. 173–213. doi:10.1021/cr60018a003.
- Clark R. Landis, Frank Weinhold: Valency and bonding: a natural bond orbital donor-acceptor perspective. Cambridge University Press, Cambridge, UK 2005, ISBN 0-521-83128-8, S. 96–100.
- K.-P. Huber, Gerhard Herzberg: Molecular Spectra and Molecular Structure. IV. Constants of Diatomic Molecules. Van Nostrand Reinhold, New York 1979.
- Jiří Cížek, Robert J. Damburg, Sandro Graffi, Vincenzo Grecchi, Evans M. Harrell, Jonathan G. Harris, Sachiko Nakai, Josef Paldus, Rafail Kh. Propin, Harris J. Silverstone: 1/R expansion for H2+: Calculation of exponentially small terms and asymptotics. In: Physical Review A. Band 33, Nr. 1, 1986, S. 12–54, doi:10.1103/PhysRevA.33.12.
- T. C. Scott, Alexander Dalgarno, J.D. Morgan III: Exchange Energy of H2+ Calculated from Polarization Perturbation Theory and the Holstein-Herring Method. In: Phys. Rev. Lett. 67, 1991, S. 1419–1422. doi:10.1103/PhysRevLett.67.1419
- G. Hadinger, M. Aubert-Frécon, G. Hadinger: The Killingbeck method for the one-electron two-centre problem. In: Journal of Physics B. 22, 1989, S. 697–712, doi:10.1088/0953-4075/22/5/003.
- E. Herbst: The Astrochemistry of H3+. In: Phil. Trans. R. Soc. Lond. A. 2000, 358, 1774, S. 2523–2534, doi:10.1098/rsta.2000.0665.