Alpha-1-Antitrypsin-Mangel
Der α1-Antitrypsin-Mangel (Synonyme: Laurell-Eriksson-Syndrom, Proteaseinhibitormangel, AAT-Defizit) ist eine erbliche Stoffwechselerkrankung aufgrund eines Polymorphismus des Proteinase-Systems. Ein Mangel an Proteaseinhibitoren kann zu Leberzirrhose und Lungenemphysem führen.
| Klassifikation nach ICD-10 | |
|---|---|
| E88.0 | Störungen des Plasmaprotein-Stoffwechsels, anderenorts nicht klassifiziert |
| ICD-10 online (WHO-Version 2019) | |
Geschichte
Antitrypsin-Mutationen verbreiteten sich möglicherweise in der Eisenzeit in Europa, da sie einen evolutionären Vorteil darstellten, indem sie die Entzündungsreaktion fokussierten und verstärkten, um invasive gastrointestinale und respiratorische Infektionen zu begrenzen. Erst seit der Entdeckung von Antibiotika sind durch die Verbreitung des Rauchens und die längere Lebensdauer diese Schutzmutationen schädlich geworden.[1]
Carl-Bertil Laurell und Sten Eriksson beschrieben die Erkrankung als erste in den frühen 60er Jahren. In einer Familie waren Emphyseme gehäuft aufgetreten.
Genetik
- Erbgang: autosomal-co-dominant[2] (AR)
- Genlokus: Chromosom 14q32.1
- Häufigkeit: 1:2000 bis 1:5000, die Erkrankung wird oft nicht erkannt
- Mechanismus: Ungehinderte Proteolyse des Gewebes durch Proteasen (v. a. aus neutrophilen Granulozyten)
Mutationen an Position 342 (PiZZ) führen immer zu klinischen Erscheinungen, Mutationen an Position 264 (PiSS) bleiben jedoch meist stumm.
| Lebenszeitrisiko nach Genotyp[3][4] | ||||
|---|---|---|---|---|
| Genotyp |
Häufigkeit in Europa |
Typische Serum- konzentration |
Emphysem- risiko |
Cirrhose- risiko |
| PiMM (normal) |
91,1 % | 20–53 μmol/L 102–254 mg/dL |
normal | normal |
| PiMS (Träger, subklinisch) |
6,6 % | 18–52 μmol/L
86–218 mg/dL |
normal | normal |
| PiMZ (Träger, mild bis moderat) |
1,9 % | 15–42 μmol/L
62–151 mg/dL |
leicht erhöht | möglich |
| PiSS (homozygot, mild) |
0,3 % | 20–48 μmol/L
43–154 mg/dL |
leicht erhöht | normal |
| PiSZ (moderat) |
0,1 % | 10–23 μmol/L
33–108 mg/dL |
leicht erhöht
bis erhöht* |
möglich |
| PiZZ (homozygot, schwer) |
0,01 % | 3,4–7 μmol/L
<29–52 mg/dL |
hoch | hoch |
| Pi00 | - | 0 μmol/L
0 mg/dL |
sehr hoch | normal |
- *Anhand empirischer Daten wurde ermittelt, dass eine Serumkonzentration <57 mg/dL mit einem erhöhten Risiko für eine Lungenkrankheit einhergeht.
Pathogenese
Das α1-Antitrypsin ist ein Akute-Phasen-Protein und einer der wichtigsten Proteinaseninhibitoren im Serum. Er hemmt u. a. die Proteinasen Trypsin und Neutrophilenelastase (NE). Ein Mangel kann zu verstärkter Proteolyse führen. Die normale Serumkonzentration beträgt 0,9–2,0 g/l.
Die Mutationen führen zu einer Strukturänderung der Proteine. Dies hat eine gestörte Sekretion und fehlerhafte Funktion zur Folge. Es kommt zu Aggregation und Akkumulation im Endoplasmatischen Retikulum (ER) der Leberzellen (Hepatozyten) und in weiterer Folge zu einem Mangel im Zellplasma (meist auf unter 40 % des Normalwertes). Das hat eine verminderte Proteinaseinhibitor-Aktivität und somit verstärkte Proteolyse zur Folge.
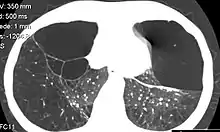
Die ungehemmte Leukozytenelastase zerstört das Lungengerüst. Es entwickelt sich ein progredientes Lungenemphysem. Die Akkumulation von α1-Antitrypsin im Endoplasmatischen Retikulum der Hepatozyten führt zu Zellschäden und in weiterer Folge zu Fibrose und Leberzirrhose.
Symptome
Bei den meisten Patienten liegt eine chronisch-aktive Leberentzündung (Hepatitis) vor, die oft schon im Kindesalter auffällig wird. Im späteren Lebensalter entwickeln bis zu 40 % der Betroffenen eine Leberzirrhose und etwa 15 % ein hepatozelluläres Carcinom (HCC).
Die bedeutendste Manifestation bei Homozygoten ist die chronisch-obstruktive Lungenerkrankung (COPD). Diese tritt meist erst nach dem 30. Lebensjahr auf. Das Lungenemphysem und die Komplikationen der COPD können über die respiratorische Insuffizienz zur Hypoxämie mit möglicher Rechtsherzinsuffizienz und Cor pulmonale zum Multiorganversagen führen. In einigen Fällen kann sich auch die Atempumpe erschöpfen und eine Hyperkapnie mit einem erhöhten arteriellen Kohlendioxidpartialdruck als Komplikation hinzukommen.
Gelegentlich kommt es zu Glomerulonephritis, nekrotisierender Vaskulitis, Granulomatose mit Polyangiitis, nekrotisierender Pannikulitis, Pankreatitis und Pankreasfibrose.
Diagnostik
Die Diagnostik ist bei allen Patienten mit COPD einmalig empfehlenswert. Das gilt insbesondere bei ungewöhnlichem Verlauf, jüngeren Personen und Nichtrauchern, die eine COPD entwickeln.[5]
Die diagnostischen Kriterien für den Nachweis eines α1-Antitrypsinmangels sind:
- α1-Antitrypsin < 0,9 g/l
- Nachweis der Genotypen PiZZ, PiMZ und PiSZ
- PAS-positive, proteaseresistente hepatozelluläre Einschlusskörperchen (=Antitrypsinablagerungen)
Oft dauert es von den ersten Beschwerden bis zur Diagnose mehrere Jahre. Viele Patienten werden zu spät entdeckt, ihnen wird nur geraten, das Rauchen einzustellen und sie werden unzureichend und ohne geeignete Untersuchungen jahrelang behandelt.
Therapie
Neben der in manchen Fällen erforderlichen Substitution werden in erster Linie die Folgeerkrankungen behandelt, vor allem die chronisch obstruktive Lungenerkrankung. Ein absoluter Rauchverzicht ist unbedingt nötig, da die im Rauch enthaltenen Oxidantien α1-Antitrypsin inaktivieren. Ebenso ist es ratsam, sich von Feuerrauch und Stäuben (Heu, Schleifarbeiten) jeglicher Art fernzuhalten oder Atemschutz zu tragen. Letzteres wird manchmal empfindlichen Personen empfohlen, um zu versuchen, sich vor Infekten zu schützen. Eine geeignete medikamentöse Behandlung ist meistens nötig, um die Folgen der COPD und besonders von Infekten möglichst gering zu halten.
In diesem Zusammenhang sind auch Impfungen empfehlenswert (Grippe, Pneumokokken).
Substitutionstherapie
Bei schwerem Lungenemphysem empfiehlt sich in manchen Fällen der intravenöse Ersatz (Substitutionstherapie) von α1-Antitrypsin. Es sollte ein Spiegel über 0,8 g/l angestrebt werden, um damit wieder in den Bereich der klinisch unauffälligen Personen zu kommen. Die Substitution bringt keinen Vorteil bei Vorliegen eines Leberschadens, weil hier die Akkumulation im Vordergrund steht.
Organtransplantation
Im fortgeschrittenen Stadium kann eine Lungen- oder Lebertransplantation nötig sein. Die Lebertransplantation ist kurativ, weil α1-Antitrypsin kaum in extrahepatischem Gewebe synthetisiert wird, behebt aber nicht die schon entstandenen Lungenschäden.
Assoziierte Erkrankungen
α1-Antitrypsin-Mangel ist mit einer Reihe von Erkrankungen assoziiert:
- Leberzirrhose
- COPD
- Pneumothorax
- Asthma
- Granulomatose mit Polyangiitis
- Pankreatitis
- Cholezystolithiasis (Gallensteine)
- Bronchiektasien
- Prolaps von Beckenorganen
- Primär sklerosierende Cholangitis
- Autoimmunhepatitis
- Emphysem, betrifft vorwiegend die Unterlappen und verursacht Bullae
- Carcinome (Krebs)
Literatur
- Alexander Biedermann, Thomas Köhnlein: Alpha-1-Antitrypsin-Mangel – eine versteckte Ursache der COPD: Überblick über Pathogenese, Diagnostik, Klinik und Therapie. Deutsches Ärzteblatt, 103. Jahrgang, Ausgabe 26 vom 30. Juni 2006, S. A-1828 / B-1569 / C-1518
- L. Fregonese, J. Stolk: Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. In: Orphanet J Rare Dis. 2008 Jun 19;3, S. 16. Review. PMID 18565211, PMC 2441617 (freier Volltext)
- E. K. Silverman, R. A. Sandhaus: Clinical practice: Alpha1-antitrypsin deficiency. In: New England Journal of Medicine. (2009) vol 360 (26), S. 2749–2757. Review. PMID 19553648
- J. Stoller, L. Aboussouan: Alpha1-antitrypsin deficiency. In: Lancet. (2005) 365 (9478), S. 2225–2236. PMID 15978931
Weblinks
Einzelnachweise
- David A. Lomas: The Selective Advantage of α -Antitrypsin Deficiency . In: American Journal of Respiratory and Critical Care Medicine. 173, 2006, S. 1072, doi:10.1164/rccm.200511-1797PP.
- Christoph Höner zu Siederdissen, Thomas Köhnlein: Alpha-1-Antitrypsinmangel. In: Praxis der Hepatologie. Springer Berlin Heidelberg, Berlin, Heidelberg 2016, ISBN 978-3-642-41620-0, S. 155–160, doi:10.1007/978-3-642-41620-0_24.
- James K. Stoller, Felicitas L. Lacbawan, Loutfi S. Aboussouan: Alpha-1 Antitrypsin Deficiency. In: GeneReviews(®). University of Washington, Seattle, Seattle (WA) 1. Januar 1993, PMID 20301692 (nih.gov [abgerufen am 1. Februar 2017]).
- Sarah K. Brode, Simon C. Ling, Kenneth R. Chapman: Alpha-1 antitrypsin deficiency: a commonly overlooked cause of lung disease. In: CMAJ : Canadian Medical Association Journal. Band 184, Nr. 12, 4. September 2012, ISSN 0820-3946, S. 1365–1371, doi:10.1503/cmaj.111749, PMID 22761482, PMC 3447047 (freier Volltext).
- Vogelmeier, C et al., Leitlinie COPD, Pneumologie 2007;61: e1-e40.