Adulte Polyglucosankörper-Erkrankung
Die adulte Polyglucosankörper-Erkrankung (englisch: adult polyglucosan body disease, abgekürzt APBD) ist eine autosomal-rezessiv vererbte Stoffwechselerkrankung, die zur Gruppe der Glykogenspeicherkrankheiten (Glykogenosen) gehört. Die sehr seltene Erkrankung wurde 1980 erstmals beschrieben[1] und macht sich klinisch vor allem durch eine langsam fortschreitende dementielle Entwicklung und Muskelschwäche bemerkbar. Auch Blasenfunktionsstörungen, eine Polyneuropathie oder eine Parkinson-Symptomatik können auftreten.
| Klassifikation nach ICD-10 | |
|---|---|
| E74.0 | Glykogenspeicherkrankheit (Glykogenose) |
| ICD-10 online (WHO-Version 2019) | |
Zum Teil geht die Erkrankung (wie die Glykogenose Typ IV) mit einer reduzierten Aktivität des 1,4-α-Glucan-verzweigenden Enzyms einher.[2] Mutationen des GBE1-Gens, das für dieses am Aufbau von Glykogen beteiligten Enzyms kodiert, konnten nachgewiesen werden.[3]
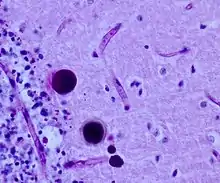
Neuropathologisch ist der Nachweis von Polyglucosankörpern charakteristisch. Hierbei handelt es sich um Polysaccharid-haltige Einschlusskörper in Astrozyten und auch Nervenzellfortsätzen, die so genannten Lafora-Körpern ähneln und sich in der PAS-Färbung kräftig markieren. PAS-positive Einschlusskörper treten auch im peripheren Nervensystem und anderen Organen auf; deren Nachweis kann zum Beispiel in der Nervenbiopsie diagnostisch sein.
Der Verlauf der Erkrankung ist langsam fortschreitend, eine spezifische Behandlung nicht möglich.
Einzelnachweise
- Y. Robitaille u. a.: A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora's disease and normal ageing. In: Brain. 1980;103(2), S. 315–336, PMID 6249438.
- C. Bruno u. a.: Glycogen branching enzyme deficiency in adult polyglucosan body disease. In: Ann Neurol. 1993;33(1), S. 88–93, PMID 8494336.
- A. Lossos u. a.: Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. In: Ann Neurol. 1998;44(6), S. 867–872, PMID 9851430.