Ösophagusatresie
Ösophagusatresie ist eine angeborene Fehlbildung, bei der eine Unterbrechung der Speiseröhre im Vordergrund steht: Entweder hat die Speiseröhre keine Verbindung zum Magen und mündet in die Luftröhre oder sie hat eine so starke Verengung (Stenose), dass keine Nahrung passieren kann. Die Häufigkeit der Ösophagusatresie liegt bei etwa 1:3500 Geburten.
| Klassifikation nach ICD-10 | |
|---|---|
| Q39.0 | Ösophagusatresie ohne Fistel |
| Q39.1 | Ösophagusatresie mit Ösophagotrachealfistel |
| ICD-10 online (WHO-Version 2019) | |
Pathogenese
Die Speiseröhre entwickelt sich während der Fetalzeit aus dem embryonalen Vorderdarm, der sich vom Pharynx bis zum Magen des Keimlings erstreckt. Ab dem 20. Schwangerschaftstag ist eine bauchseitige Verdickung nachweisbar, aus der sich das respiratorische Zylinderepithel differenziert, welches ab dem 26. Schwangerschaftstag eine vollständige Separation in Form einer neuen Röhre – der späteren Luftröhre – ausbildet. Ist dieser Separationsvorgang gestört, entwickelt sich eine Ösophagusatresie.
Einteilung
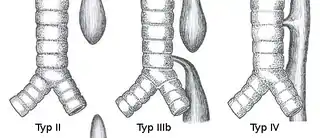
Die verschiedenen Formen der Ösophagusatresie werden nach Vogt folgendermaßen eingeteilt:
- Vogt Typ I: Ösophagusaplasie (keine Luftansammlung im Magen), Häufigkeit ca. 1 %
- Vogt Typ II: Atresie ohne ösophagotracheale Fistelbildung (ebenfalls keine Luftansammlung im Magen), Häufigkeit ca. 6 %
- Vogt Typ IIIa: ösophagotracheale Fistel am oberen Segment, das untere Segment endet im Blindsack, Häufigkeit ca. 1 %
- Vogt Typ IIIb: ösophagotracheale Fistelbildung am unteren Segment, das obere Segment endet im Blindsack, Häufigkeit ca. 85 %
- Vogt Typ IIIc: ösophagotracheale Fistelbildung am unteren und oberen Segment, Häufigkeit ca. 5 %
- Vogt Typ IV: Tracheo-ösophageale Fistel ohne Atresie (sog. „H-Fistel“), Häufigkeit: ca. 2 %.
Die Angaben zur Häufigkeit schwanken von Autor zu Autor.
Das äußerst seltene vollständige Fehlen der Speiseröhre wird als Ösophagusagenesie bezeichnet.
Symptomatik
Die neugeborenen Patienten – häufig sind es Frühgeborene – fallen durch Husten, Speicheln und Verschlechterung ihres Allgemeinzustandes auf. Häufig laufen große Mengen schaumigen Speichels aus dem Mund, ohne dass sie geschluckt werden können. Teilweise wird der Speichel sogar herausgewürgt, auch Hustenattacken und Zyanoseanfälle können beim Kind auftreten. Nach einem Fütterungsversuch entwickelt sich durch Nahrungsaspiration eine Zyanose. Bei Vogt IV leiden die erkrankten Säuglinge an wiederholten Aspirationspneumonien, ohne weitere Symptome aufzuzeigen.
Begleitfehlbildung
In manchen Fällen ist eine Oesophagusatresie mit einem Herzfehler kombiniert. Weitere typische Fehlbildungen sind solche der Extremitäten, Wirbelkörperanomalien, Nierenfehlbildung oder Darmatresien zum Beispiel im Rahmen einer VACTERL-Assoziation.
Diagnostik
Vorgeburtlich weist der sonografische Softmarker eines Polyhydramnions (zu viel Fruchtwasser) auf eine mögliche Ösophagusatresie hin.
Nach der Geburt weist die Symptomatik auf eine Oesophagusatresie hin.
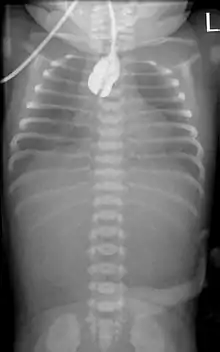
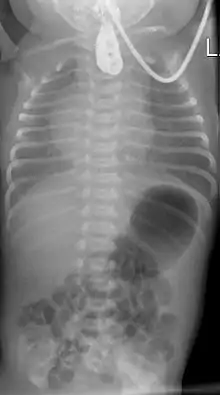
Zur Diagnosefindung wird die Speiseröhre sondiert. Ein federnder Stopp ist hinweisend. Eine Röntgenaufnahme des Brustkorbes zeigt die Luftfüllung des oberen Blindsackes (sog. Medaillonzeichen), und gegebenenfalls eine Luftfüllung des Darmes als Hinweis auf eine untere Fistel. Nur in Ausnahmefällen wird zusätzlich wasserlösliches Kontrastmittel gegeben.
Weitere Fehlbildungen müssen mittels Ultraschall gesucht werden. Zur Operationsplanung muss im Echokardiogramm die Lage der Hauptschlagader dargestellt werden.
Therapie
Die Operation einer angeborenen (kongenitalen) Ösophagusatresie beim Neugeborenen gelang erstmals Cameron Haight in Ann Arbor.[1]
Vor einer Operation wird der Oberkörper des Kindes hochgelagert und das Sekret aus dem nicht durchgängigen Teil konstant durch eine Sonde abgesaugt.
Die operative Korrektur richtet sich nach dem Abstand des oberen Speiseröhrenanteils zum unteren. Ist der Abstand gering, können mit einer Operation, die rasch erfolgt, beide Oesophagusanteile miteinander verbunden werden. Ist der Abstand groß, müssen alternative Therapien erfolgen. Es erfolgt entweder eine Verlängerungsbehandlung des Ösophagus über mehrere Tage oder Wochen, bis die Distanz kurz genug ist oder die fehlende Speiseröhre wird durch Magen- oder Darmanteile, die in den Brustkorb verlagert werden, ersetzt. Besteht eine Verbindung zur Luftröhre oder Lunge, muss diese in jedem Fall operativ durchtrennt und verschlossen werden, um zu verhindern, dass Speichel oder Nahrung in die Atemwege gelangt.
Verlauf und Prognose
Eine Nachbehandlung ist in der Regel über mehrere Jahre notwendig. Typische Probleme sind Tracheomalazie, Gastroösophagealer Reflux, Stenosen an der Ösophagusnahtstelle. Die Letalität ist abhängig von Begleitfehlbildungen und vom Geburtsgewicht. Sie liegt bei einem Geburtsgewicht über 1500 g und ohne wesentliche Herzfehler unter 5 %.[2]
Literatur
- Lewis Spitz: Oesophageal atresia. In: Orphanet Journal of Rare Diseases. Band 2, Nr. 1, Mai 2007, ISSN 1750-1172, S. 24, doi:10.1186/1750-1172-2-24 (englisch).
- Leitlinien
- S2k-Leitlinie Kurzstreckige Ösophagusatresie der Deutschen Gesellschaft für Kinderchirurgie (DGKCH). In: AWMF online (Stand 2012)
Weblinks
Einzelnachweise
- Ernst Kern: Sehen – Denken – Handeln eines Chirurgen im 20. Jahrhundert. ecomed, Landsberg am Lech 2000. ISBN 3-609-20149-5, S. 300.
- M. Yagyu, H. Gitter, B. Richter, D. Booss: Esophageal atresia in Bremen, Germany–evaluation of preoperative risk classification in esophageal atresia. In: Journal of pediatric surgery. Band 35, Nummer 4, April 2000, S. 584–587, ISSN 0022-3468. doi:10.1053/jpsu.2000.0350584. PMID 10770387.