Rosai-Dorfman-Erkrankung
Das Rosai-Dorfman-Syndrom ist eine seltene Erkrankung aus der Gruppe der Histiozytosen. Die Erkrankung ist charakterisiert durch eine Proliferation von Histiozyten, welche sich überwiegend in den Lymphknoten ansammeln.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| D76.3 | Sonstige Histiozytosesyndrome |
| ICD-10 online (WHO-Version 2019) | |
Synonyme: Sinus-Histiozytose Rosai Dorfman; Sinus-Histiozytose mit massiver Lymphadenopathie; Histiozytische lymphophagozytische Pannikulitis; Benigne Pseudolymphomatose; 'Destombes-Rosaï-Dorfman-Krankheit; Rosaï-Dorfman-Destombes-Krankheit; SHML
Die Erstbeschreibung erfolgte im Jahre 1969 durch die US-amerikanischen Pathologen Juan Rosai und Ronald F. Dorfman.[3]
Pathologie
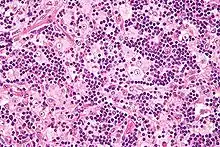
Es zeigt sich eine ausgeprägte, beidseitige Vergrößerung der zervikalen Lymphknoten, die hauptsächlich auf die Verbreiterung der Sinus der Lymphknoten zurückzuführen ist. Diese Sinus sind mit aktivierten Histiozyten gefüllt, die intakte Erythrozyten und Lymphozyten umhüllen, was als Emperipolese bezeichnet wird.[4][5]
Gleichzeitig findet in den Sinus eine Vermehrung von Plasmazellen, Lymphozyten und neutrophilen Granulozyten statt. Auch andere Lokalisationen sind möglich: so können Herde in der Haut, der Hirnhaut, dem oberen Respirationstrakt, Speicheldrüsen und Knochenmark auftreten.[5]
Vorkommen
Die Häufigkeit ist bislang ungeklärt, man nimmt an, dass Virusinfektionen eine Rolle spielen können. In Europa ist das Rosai-Dorfman-Syndrom sehr selten und betrifft v. a. Jugendliche im Alter von 15 bis 20 Jahren, weltweit sind häufiger Menschen schwarzer Hautfarbe betroffen.
Ursache
Der Erkrankung liegen Mutationen im SLC29A33-Gen zugrunde, welches für einen Nukleosid-Transporter kodiert.
Das gleiche Gen ist auch beim H-Syndrom und der Dysosteosklerose betroffen.[6][1]
Klinische Erscheinungen
Neben ausgeprägten Lymphknotenschwellungen (Lymphadenopathie) können Allgemeinsymptome wie Fieber und Gewichtsverlust auftreten.[7][8]
In etwa der Hälfte der Fälle finden sich Krankheitsherde außerhalb der Lymphknoten (Haut, oberer Respirationstrakt, Speicheldrüsen), die sich dann durch spezifische Symptome für den Lokalisationsort zeigen.
Der Krankheitsverlauf ist überwiegend selbstlimitierend, laborchemisch finden sich allgemeine Entzündungszeichen.[8]
Literatur
- S. Vandersee, H. J. Röwert-Huber, S. Wöhner, C. Loddenkemper, M. Beyer, D. Humme: Kutanes Rosai-Dorfman-Syndrom. Erfolgreiche Therapie mit intraläsional applizierten Steroiden. In: Der Hautarzt; Zeitschrift für Dermatologie, Venerologie, und verwandte Gebiete. Band 65, Nr. 8, August 2014, S. 725–727, doi:10.1007/s00105-014-2797-0, PMID 24831530.
- Werner Böcker: Lehrbuch Pathologie. 4. Auflage. Urban & Fischer Verlag, 2008, ISBN 978-3-437-42382-6
Einzelnachweise
- Rosaï-Dorfman-Krankheit. In: Orphanet (Datenbank für seltene Krankheiten).
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- J. Rosai, R. F. Dorfman: Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. In: Archives of pathology. Band 87, Nummer 1, Januar 1969, S. 63–70, PMID 5782438.
- W. D. James, T. G. Berger, D. M. Elston: Andrews’ Diseases of the Skin: Clinical Dermatology. 11. Auflage. Saunders Elsevier, 2006, ISBN 978-1-4377-0314-6
- E. Foucar, J. Rosai, R. Dorfman: Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. In: Seminars in diagnostic pathology. Band 7, Nr. 1, Februar 1990, S. 19–73, PMID 2180012 (Review).
- Dysosteosclerosis. In: Online Mendelian Inheritance in Man. (englisch)
- W. Hindermann, D. Katenkamp: Extranodale Rosai-Dorfman-Erkrankung (Sinushistiozytose mit massiver Lymphadenopathie). In: Der Pathologe. 25, 2004, S. 222, doi:10.1007/s00292-003-0642-9
- Werner Böcker et al. (Hrsg.): Lehrbuch Pathologie 4. Auflage. Urban & Fischer Verlag, 2008, ISBN 978-3-437-42382-6