Quantitative Trait Locus
Als Quantitative Trait Locus (abgekürzt QTL, Mehrzahl Quantitative Trait Loci, deutsch: Region eines quantitativen Merkmals) wird in der Genetik ein Abschnitt eines Chromosoms bezeichnet, für den in entsprechenden Studien ein Einfluss auf die Ausprägung eines quantitativen phänotypischen Merkmals des betreffenden Organismus nachgewiesen wurde. Während diskrete Merkmale, wie beispielsweise die Blütenfarbe bei Pflanzen, in mehreren verschiedenen, voneinander abgegrenzten Zuständen vorliegen, sind quantitative (stetige) Merkmale wie zum Beispiel die Körpergröße und das Körpergewicht ohne Abstufung auf einer kontinuierlichen Skala messbar. Die Identifizierung von Chromosomenabschnitten mit einem Einfluss auf die Ausprägung solcher Merkmale ist vor allem in der Humangenetik zum Auffinden von krankheitsrelevanten Genen, in der landwirtschaftlichen Züchtungsforschung und in der Evolutionsbiologie von Interesse. Studien zur Identifizierung von QTL werden seit etwa 1990 durchgeführt.
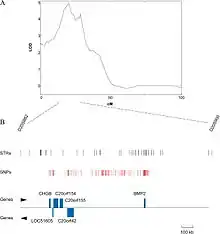
Bestimmung von Quantitative Trait Loci
Zur Analyse von QTL wird in Studien mit einer großen Zahl an individuellen Lebewesen die Ausprägung eines bestimmten Merkmals in Relation zu Variationen von genetischen Markern untersucht. Diese Marker sind Abschnitte in der DNA-Sequenz, die in verschiedenen Individuen in unterschiedlichen Formen, sogenannten Allelen, auftreten. Für QTL-Analysen sind vor allem Genmarker mit einem hohen Polymorphismusgrad, also einer hohen Zahl an verschiedenen Allelen, von Interesse. Dabei werden RFLP-, SNP- und STR-Polymorphismen besonders häufig als Marker genutzt. Alle diese Polymorphismen sind sogenannte anonyme Marker, da ihnen keine funktionelle Bedeutung zugeordnet ist und sie im Regelfall keinem spezifischen Gen entsprechen.
Für sogenannte genomweite QTL-Studien wird eine größere Zahl an Markern ausgewählt, die gleichmäßig über das gesamte Genom der zu untersuchenden Lebewesen verteilt sind. Die Zahl der Marker in Relation zur gesamten Genomgröße wird als Markerdichte bezeichnet, eine höhere Markerdichte erlaubt dabei eine bessere Eingrenzung der QTL auf einen kleineren chromosomalen Bereich. Basierend auf einer solchen Markerauswahl sind zwei verschiedene Untersuchungsansätze möglich.
Assoziationsstudien
In sogenannten genomweiten Assoziationsstudien werden in allen Individuen einer Studiengruppe für jeden Marker die entsprechenden Allele bestimmt. Unter der Annahme, dass ein bestimmter Marker in keiner Beziehung zum untersuchten quantitativen Merkmal steht, wird von einer vergleichbaren Verteilung der Allele in allen Bereichen der Merkmalsausprägung ausgegangen. Die Häufigkeit der einzelnen Allele eines Markers unterscheidet sich in diesem Fall also nicht zwischen Individuengruppen mit Unterschieden in der Merkmalsausprägung.
Eine zu- oder abnehmende Häufigkeit von bestimmten Allelen eines Markers mit zu- oder abnehmender Ausprägung des untersuchten Merkmals, oder eine anderweitig markant unterschiedliche Allelverteilung eines Markers in verschiedenen Bereichen der Merkmalsausprägung, also beispielsweise den großen und den kleinen Individuen der untersuchten Gruppe, ist hingegen ein Hinweis auf eine Assoziation dieses Markers zum untersuchten Merkmal. Dies liegt daran, dass mit abnehmendem Abstand eines Markers zu einem Gen, das die Merkmalsausprägung beeinflusst, die Wahrscheinlichkeit steigt, dass dieser Marker mit diesem Gen gekoppelt vererbt wird. Dies hat zur Folge, dass die Allelverteilung dieses Markers nicht mehr zufällig, sondern mit der Merkmalsausprägung assoziiert ist. Die Wahrscheinlichkeit dafür wird durch einen sogenannten LOD-Score ermittelt. Ein LOD-Score von größer als drei für einen Marker wird in QTL-Studien im Allgemeinen als Indikator für einen QTL an der Position dieses Markers angesehen.
Häufiger werden, analog zum beschriebenen Ansatz, zwei Studiengruppen untersucht, die sich in der Merkmalsausprägung markant unterscheiden, beispielsweise eine Züchtungsform einer Pflanzenart mit normaler Wuchshöhe und eine weitere mit Zwergwuchs, oder eine Gruppe von Patienten mit einer bestimmten Erkrankung und eine weitere Gruppen von gesunden Personen. In diesem Fall wird für jeden Marker die Allelhäufigkeit in beiden Gruppen bestimmt. Marker mit ausgeprägt unterschiedlicher Allelverteilung zwischen beiden Gruppen weisen dann eine Assoziation zum untersuchten Merkmal auf. Die beiden untersuchten Studiengruppen sollten sich allerdings mit Ausnahme des zu untersuchenden Merkmals in Phänotyp und Genotyp möglichst ähnlich sein. Für Assoziationsstudien wird je nach Relevanz der zu untersuchenden Marker für die Merkmalsausprägung eine bis zu drei- oder vierstellige Zahl an Individuen benötigt. Sie werden deshalb insbesondere im Bereich der Züchtungsforschung an Pflanzen und Kleintieren durchgeführt.
Kopplungsstudien

In Kopplungsstudien wird bei erstgradigen Verwandten, also entweder zwischen Eltern und ihren Kindern oder zwischen Geschwistern, die sich hinsichtlich des zu untersuchenden Merkmals ähnlich sind, die Vererbung der ausgewählten Marker untersucht. Wenn ein Marker keinen Bezug zum untersuchten Merkmal aufweist, sind ein Elternteil und ein Kind unter der Annahme einer zufälligen Vererbung in 50 Prozent aller untersuchten Fälle identisch (konkordant) hinsichtlich ihres jeweiligen Allels dieses Markers. Je dichter sich jedoch ein Marker an einem Gen befindet, welches das untersuchte Merkmal beeinflusst, umso mehr weicht die Häufigkeit von konkordanten Eltern-Kind-Kombinationen von der aus einer zufälligen Vererbung resultierenden Wahrscheinlichkeit von 50 Prozent ab. Dies wird als Transmission disequilibrium (Vererbungsungleichgewicht) oder Kopplungsungleichgewicht bezeichnet.
Analog dazu ist bei zwei Geschwistern die aufgrund einer zufälligen Vererbung zu erwartende Verteilung, dass 25 Prozent aller Geschwisterpaare für einen bestimmten Marker kein gemeinsames Allel besitzen, in 50 Prozent aller Fälle ein gemeinsames Allel aufweisen und in 25 Prozent aller Fälle in beiden Allelen übereinstimmen. Auch hier zeigt eine Abweichung von dieser Verteilung für einen bestimmten Marker in einer entsprechenden Zahl von untersuchten Geschwisterpaaren eine Nähe dieses Markers zu einem Gen an, das einen Einfluss auf das untersuchte Merkmal hat. Dieses jeweilige Gen und der Marker sind gekoppelt.
Kopplungsstudien sind insbesondere von Relevanz, wenn für Assoziationsstudien keine ausreichend große Zahl an Individuen zur Verfügung steht, wenn der Einfluss der einzelnen QTL auf die Merkmalsausprägung nur gering ist oder wenn die zu untersuchenden Marker nur wenige verschiedene Allele aufweisen. Sie sind deshalb insbesondere in der Humangenetik von Relevanz.
Ein weiteres ebenfalls auf der Analyse von Geschwisterpaaren beruhendes Konzept wird als Affected Family based Control (AFBAC) bezeichnet. Dabei wird eine Gruppe von Geschwisterpaaren untersucht, die hinsichtlich des betreffenden Merkmals diskordant sind, sich also in diesem Merkmal in zwei verschiedenen Zuständen unterscheiden. Dies können beispielsweise Geschwisterpaare mit einer normalgewichtigen und einer übergewichtigen Person sein. Weicht hier die Allelverteilung eines Markers bei den Personen mit der einen Variante der Merkmalsausprägung von den Personen mit der anderen Variante ab, ist dies ein Hinweis auf einen Zusammenhang zwischen diesem Marker und der Merkmalsausprägung. Auch wenn AFBAC-Studien auf der Untersuchung von erstgradig verwandten Personen beruhen, zählen sie zu den Assoziationsstudien, da nicht die Vererbung untersucht wird. Beim Vorliegen einer ausreichend großen Zahl an vollständig untersuchten Familien ist jedoch auch die Kombination von AFBAC- und Kopplungsuntersuchungen innerhalb der gleichen Studiengruppe möglich.
Relevanz von Quantitative Trait Loci
Für diskrete Merkmale ist die Identifizierung der Gene, die für ihre Vererbung verantwortlich sind, in vielen Fällen vergleichsweise einfach. In der Regel sind nur ein oder wenige Gene an der Ausprägung solcher Merkmale beteiligt, die Vererbung entspricht damit oft einem klassischen Erbgang oder kann durch einen solchen hinreichend genau beschrieben werden. Im Gegensatz dazu wird die Ausprägung von quantitativen Merkmalen fast immer durch Interaktionen einer größeren Zahl an Genen sowie durch zusätzliche Wechselwirkungen der genetischen Veranlagung mit Umweltfaktoren bestimmt. Eine solche als „polygenetisch“ bezeichnete Vererbung folgt keinem klassischen Erbgang, da der Anteil jedes einzelnen Gens an der Merkmalsausprägung vergleichsweise gering ist und darüber hinaus von den genannten Wechselwirkungen abhängt. Die Identifizierung der an der Ausprägung eines quantitativen Merkmals beteiligten Gene ist deshalb methodisch sehr aufwändig.
Das Erkennen von Quantitative Trait Loci ist der erste Schritt im Rahmen der Identifizierung von Genen für quantitative Merkmale. Durch die Bestimmung von QTL werden dabei chromosomale Abschnitte bestimmt, in denen sich mögliche Kandidatengene befinden. Die Größe der QTL und damit die Zahl der Kandidatengene ergibt sich dabei aus der Markerdichte. Durch eine gezielte Untersuchung eines gefundenen Quantitative Trait Locus mit einer höheren Markerdichte ist eine weitere Eingrenzung auf einzelne Gene möglich. Die Zahl der Kandidatengene kann darüber hinaus weiter eingeschränkt werden, indem in entsprechenden Datenbanken die bekannten Gene ermittelt werden, die im gefundenen QTL liegen, und hinsichtlich eines möglichen funktionalen Bezugs zum untersuchten Merkmal bewertet werden.
Quantitative Trait Loci spielen vorrangig in der Humangenetik bei der Ermittlung von krankheitsrelevanten Genen und Genvariationen eine Rolle. Sie sind darüber hinaus auch, ohne Identifizierung der verantwortlichen Gene, in der Züchtungsforschung von Interesse. Dabei werden identifizierte QTL genutzt, um bei der Züchtung von Nutzpflanzen und Nutztieren für Kreuzungen gezielt Kreuzungspartner oder Nachkommen mit erwünschter Merkmalsausprägung auszuwählen.
Literatur
- J. I. Weller: Quantitative Trait Loci Analysis in Animals. CABI Publishing, Wallingford 2001, ISBN 0-85199-402-4
- N. J. Camp, A. Cox: Quantitative Trait Loci: Methods and Protocols. Reihe: Methods in Molecular Biology. Band 195. Humana Press, Totowa 2002, ISBN 0-89603-927-7
- A. H. Paterson: Molecular Dissection of Quantitative Traits: Progress and Prospects. In: Genome Research. 5/1995. Cold Spring Harbor Laboratory Press, S. 321–333, ISSN 1088-9051
- T. F. Mackay: The Genetic Architecture of Quantitative Traits. In: Annual Review of Genetics. 35/2001. Annual Reviews, S. 303–339, ISSN 0066-4197