Kortikobasale Degeneration
Die kortikobasale (oder corticobasale) Degeneration (CBD) ist eine langsam fortschreitende neurodegenerative Erkrankung mit Ansammlung hyperphosphorylierten Tau-Proteins im Gehirn (Tauopathie). Hauptsymptome sind Parkinson-Symptome und Nachlassen der kognitiven Fähigkeiten.
| Klassifikation nach ICD-10 | |
|---|---|
| G31.0 | Umschriebene Hirnatrophie |
| ICD-10 online (WHO-Version 2019) | |
Einordnung
Atypische Parkinson-Syndrome sind Erkrankungen, die leicht mit der Parkinson-Krankheit zu verwechseln sind. Die kortikobasale Degeneration ist nach der Multisystematrophie (MSA) und der progressiven supranukleären Parese (PSP) das dritthäufigste atypische Parkinson-Syndrom.
Häufigkeit und Ursachen
Die CBD ist eine seltene Erkrankung. Geschätzt wird, dass weniger als 1 von 100.000 Menschen daran erkrankt. Als einer der Auslöser konnte das für das Tau-Protein codierende MAPT-Gen identifiziert werden.[1]
Symptome / Diagnostik
Zum einen dominieren komplexe kortikale (die Hirnrinde betreffende) Symptome wie Apraxie. Es kommt zum kognitiven Leistungsverfall, der in eine subkortikale Demenz mündet.[2] Die Muskeleigenreflexe sind typischerweise gesteigert und Myoklonien kommen vor. Als sehr charakteristisches Zeichen wird ein Fremdheitserleben der eigenen Extremitäten (alien limb) beschrieben.
Unter den extrapyramidal-motorischen Symptomen sind meist einseitig beginnende Parkinson-Symptome wie Rigor und Hypokinese, seltener ein Ruhetremor festzustellen. Es kommt zu Dystonien von Nacken oder Extremitäten und zu einer schweren Gangstörung.
Fakultative Symptome sind Augenbewegungsstörungen und oft im Rahmen der Demenz zu wertende affektive Störung wie Depression oder generalisierte Angst.
Die Diagnose wird anhand der Symptome („klinisch“) gestellt. Zu ihrer Sicherung wäre eine feingewebliche (histologische) Untersuchung des Gehirns notwendig. Da sich aber für die Behandlung des Patienten keine ausreichende Konsequenz ergibt, wird eine Biopsie zu Lebzeiten nicht durchgeführt. In spezialisierten Zentren ist eine weitere Zuordnung mittels spezieller SPECT möglich. Dies hat aber praktisch nur Sinn im Rahmen der Forschung und nicht für die Versorgung der Patienten.
Neuropathologie
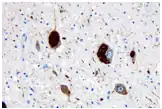
Bei der Untersuchung des Gehirns ist neuropathologisch eine oft asymmetrische Atrophie der Großhirnrinde, insbesondere der Regiones praecentralis und postcentralis nachweisbar. Die Substantia nigra ist abgeblasst. Mikroskopisch erscheint die betroffene Hirnrinde verschmälert und weist Nervenzelluntergänge auf. Typisch sind ballonierte Nervenzellen. Immunhistochemisch sind Ablagerungen von hyperphosphoryliertem Tau-Protein in Nerven- und Gliazellen nachweisbar. Neben neurofibrillären Tangles finden sich Neuropilfäden, oligodendrogliale „coiled bodies“ und astrozytäre Plaques.[3]
Behandlung, Verlauf
Die Erkrankung kann nicht geheilt oder zum Stillstand gebracht werden. Levodopa führt manchmal zu einer Besserung der Parkinson-Symptome, das Ansprechen ist jedoch meist unbefriedigend. Der klinische Verlauf ist in der Regel von zunehmenden Bewegungsstörungen mit Akinesie geprägt. Von der Diagnosestellung bis zum Tod vergehen zwischen 1 und 10 Jahre; als Todesursache findet sich häufig eine (durch Bewegungsarmut begünstigte) Lungenentzündung.[4]
Einzelnachweise
- UniProt P10636
- T. H. Bak, L. M. Crawford, V. C. Hearn, P. S. Mathuranath, J. R. Hodges: Subcortical dementia revisited: similarities and differences in cognitive function between progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and multiple system atrophy (MSA). In: Neurocase. 2005;11(4), S. 268–273.
- M. Tolnay, W. Paulus: Systematrophien. In: W. Paulus, J. M. Schröder (Hrsg.): Neuropathologie. 3. Auflage. Springer Verlag, Heidelberg 2012, ISBN 978-3-642-02323-1.
- G. K. Wenning, I. Litvan, J. Jankovic, R. Granata, C. A. Mangone, A. McKee, W. Poewe, K. Jellinger, K. Ray Chaudhuri, L. D'Olhaberriague, R. K. Pearce: Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. In: Journal of Neurology, Neurosurgery, and Psychiatry. Band 64, Nummer 2, Februar 1998, S. 184–189, ISSN 0022-3050. PMID 9489528. PMC 2169933 (freier Volltext).