Crouzon-Syndrom
Das Crouzon-Syndrom (Morbus Crouzon), Synonym: Dysostosis craniofacialis Crouzon, beschreibt eine genetische Erkrankung, die eine vorzeitige Verknöcherung der Schädelnähte (Kraniosynostose) bei Kindern auslöst. Das Kopfwachstum erfolgt dann in Richtung der noch verbleibenden offenen Schädelnähte, was zu einer Deformierung des Schädels führen kann. Typisch sind auch lateral abfallende Lidachsen. Der autosomal-dominant vererbte Morbus Crouzon wird in einer von 25.000 Geburten beschrieben und kann durch mehrere Operationen verbessert werden. Die geistige Entwicklung der betroffenen Kinder verläuft meist normal. Die Erkrankung wird nach ihrem Erstbeschreiber, dem französischen Arzt Louis Edouard Octave Crouzon benannt.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| Q75.1 | Crouzon-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Ursache
Das Crouzon-Syndrom wird durch eine autosomal-dominante Veränderung (Mutation) am langen Arm des Chromosom 10 (10q26) in einem Gen, das für den sogenannten Fibroblasten-Growth-Factor-Rezeptor 2 (FGFR2) kodiert, verursacht.[2]
Krankheitserscheinungen
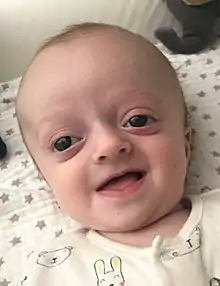
Das Crouzon-Syndrom zeichnet sich klinisch durch
- Schädeldeformitäten, Turmschädel
- Vorspringende Schädelnähte
- Exophthalmus (Hervortreten der Augäpfel)
- Hypertelorismus (vergrößerter Augenabstand), prominente Stirn
- Strabismus divergens (Schielen)
- maxilläre Retrognathie (Oberkieferhypoplasie)
- vorstehende Unterlippe
aus.
Assoziiert sind:
- Humero-radiale-Synostose (10–20 %)
- Subluxation im Ellenbogengelenk
Diagnostik
Die genannten klinischen Zeichen zusammen mit der auffälligen Schädelform und tastbaren Nähten bedürfen im Regelfall keiner bildgebenden Diagnostik. Im Röntgenbild finden sich vermehrte Impressiones digitatae sowie eine abnorme Steilstellung und Verkürzung der Schädelbasis.[3]
Differentialdiagnostik
Es gibt Formen ohne familiäre Häufung sowie ohne eine mandibuläre Prognathie, die von einigen Autoren als Pseudo-Crouzon-Syndrom abgetrennt werden.
Literatur
- Friedrich Carl Sitzmann (Hrsg.): Pädiatrie (= Duale Reihe.). 3., überarbeitete und erweiterte Auflage. Georg Thieme, Stuttgart 2007, ISBN 978-3-13-125333-0.
- B. Leiber: Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Herausgegeben von G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. Band 2, 7. Auflage. Urban & Schwarzenberg 1990, ISBN 3-541-01727-9.
Einzelnachweise
- whonamedit
- Crouzon-Syndrom. In: Online Mendelian Inheritance in Man. (englisch).
- W. Schuster, D. Färber (Hrsg.): Kinderradiologie. Bildgebende Diagnostik. Springer 1996, ISBN 3-540-60224-0.