Chondrodysplasia punctata
Unter dem Begriff Chondrodysplasia punctata wird eine Gruppe von zu den Chondrodysplasien zählender angeborener Erkrankungen zusammengefasst, die zu punktförmigen Verkalkungen (Kalzifizierungen) von Knorpelgewebe an Gelenken, Kehlkopf (Larynx) und Luftröhre (Trachea) führen.
| Klassifikation nach ICD-10 | |
|---|---|
| Q77.3 | Chondrodysplasia-punctata Syndrome |
| ICD-10 online (WHO-Version 2019) | |
Weitere Symptome sind dysproportionierter Kleinwuchs mit verkürzten Extremitäten, Verhornungsstörung der Haut, grauer Star und Dysmorphien (hypoplastische Nasenwurzel). Mitunter finden sich auch Spaltwirbel.
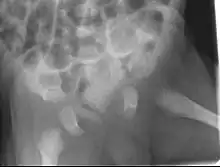
Zu der Gruppe gehören:
- Conradi-Hünermann-Syndrom: X-chromosomal-dominant vererbter Defekt des EBP-Gen (Enzym im Cholesterin-Stoffwechsel)
- Chondrodysplasie mit Brachytelephalangie: X-chromosomal-rezessiv vererbter Defekt im ARSE-Gen; eine Phänokopie dieser Erkrankung ist das Warfarin-Syndrom (Cumarin Embryopathie)[1]
- Chondrodysplasia punctata Typ Sheffield autosomal-dominant[2][3]
- Chondrodysplasia punctata, tibial-metakarpaler Typ, autosomal-dominant[4]
- Chondrodysplasia punctata Typ Toriello, autosomal-rezessiv vererbt[5]
- Chondrodyplasia punctata, rhizomeler Typ autosomal-rezessiv[6]
- Chondrodysplasia punctata, nicht-rhizomeler Typ[7]
- Chondrodysplasia punctata durch X-chromosomale Deletion[8]
Radiologisch sind die Verkalkungen lediglich bis zu einem Alter von 3 bis 5 Jahren nachweisbar. Später stehen Wachstumsverzögerungen oder -störungen im Vordergrund.
Abzugrenzen sind Knorpelverkalkungen anderer Ursache sowie das CODAS-Syndrom.
Geschichtliches
Ursprünglich wurden die Begriffe Chondrodysplasia punctata und Conradi-Hünermann synonym verwendet nach den ersten Beschreibungen durch die Kölner Kinderärzte Erich Conradi 1914 und Carl Hünermann 1931. Die heute gebräuchliche Unterscheidung beruht auf einer Abgrenzung durch Jürgen Spranger 1971.[9]
Literatur
- J. W. Spranger: Bone Dysplasias. Urban & Fischer, 2002, ISBN 3-437-21430-6.
- B. Leiber: Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Herausgegeben von G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7. Auflage. Urban & Schwarzenberg, 1990, ISBN 3-541-01727-9.
- F. Hefti: Kinderorthopädie in der Praxis. Springer, 1998, ISBN 3-540-61480-X.
Einzelnachweise
- Chondrodysplasia punctata, brachytelephalangealer Typ. In: Orphanet (Datenbank für seltene Krankheiten).
- L. Sheffield, D. Danks, V. Mayne, L. Hutchinson: Chondrodysplasia punctata - 23 cases of a mild and relatively common variety. In: Journal of Pediatrics. 1976, 89, S. 916–923.
- Chondrodysplasia punctata Typ Sheffield. In: Orphanet (Datenbank für seltene Krankheiten).
- Chondrodysplasia punctata, tibial-metakarpaler Typ. In: Orphanet (Datenbank für seltene Krankheiten).
- Chondrodysplasia punctata Typ Toriello. In: Orphanet (Datenbank für seltene Krankheiten).
- Chondrodyplasia punctata, rhizomeler Typ. In: Orphanet (Datenbank für seltene Krankheiten).
- Chondrodysplasia punctata, nicht-rhizomeler Typ. In: Orphanet (Datenbank für seltene Krankheiten).
- C. Curry, R. Magenis, M. Brown u. a.: Inherited chondrodysplasia punctata due to a deletion of the terminal short arm of an X-chromosome. In: New England Journal of Medicine. 1984, 311, S. 1010–1015.
- J. W. Spranger, J. M. Opitz, U. Bidder: Heterogenity of chondrodysplasia punctata. In: Humangenetik. 1971 11, S. 190–212.