Alveolarproteinose
Die Alveolarproteinose wird auch als pulmonal-alveoläre Proteinose oder als Phospholipoproteinose bezeichnet. Die englische Bezeichnung lautet pulmonary alveolar proteinosis (PAP). Die Erkrankung ist selten und hat eine großenteils unbekannte Ätiologie. Es liegt eine Füllung des Alveolarraums mit zellfreiem, amorphem, eosinophilem, lipidreichem Material vor, bedingt durch eine Störung des Surfactantmechanismus. Männer erkranken häufiger als Frauen, meist im mittleren Erwachsenenalter. Die Erkrankung kann klinisch unerkannt auftreten, Spontanremissionen sind möglich. Sie tritt kongenital, im Kindesalter, oder auch im mittleren Erwachsenenalter auf.
| Klassifikation nach ICD-10 | |
|---|---|
| J84.0 | Alveoläre und parietoalveoläre Krankheitszustände Alveolarproteinose |
| ICD-10 online (WHO-Version 2019) | |
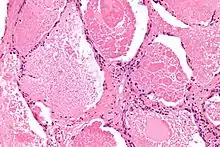
Die Alveolarproteinose wurde 1958 zuerst beschrieben[1] von den Ärzten Samuel Rosen, Benjamin Castleman, und Averill Abraham Liebow.[2]
Krankheitsbild
Die häufigen Hauptsymptome sind Luftnot und Husten. Bei der Auskultation der Lunge sind Rasselgeräusche hörbar. Erstaunlicherweise ist der Husten unproduktiv, es wird nur wenig abgehustet. Gewichtsverlust und Schwäche sind häufig, in schweren Fällen wird eine Zyanose beobachtet. Beim Erwachsenen beginnt die Erkrankung über mehrere Wochen oder Monate mit langsam zunehmenden Beschwerden. Weil das proteinhaltige Sekret in den Alveolen einen guten Nährboden darstellt, kommt es manchmal zu einer bakteriellen oder mykotischen Lungenentzündung.
Ätiologie und Pathogenese
Ursächlich für die Erkrankung sind Stoffwechselstörungen, die zur Anhäufung von Surfactant führen, indem eine pathologische Wiederaufnahme der Phospholipide über den Alveolarfilm besteht und die Phagozytosefähigkeit der Alveolarmakrophagen überlastet ist. Angeborene Formen gehen auf genetische Neumutationen zurück und sind in der Regel im Säuglingsalter tödlich. Beschrieben sind Mutationen des Surfactant-Protein-B-Gens sowie für GM-CSF. Erworbene Störungen des Surfactantmetabolismus werden als sekundäre Alveolarproteinosen bezeichnet. Sie entstehen bei pulmonalen Infekten, immunologischen Erkrankungen. Bei Tumorerkrankungen und auch chronischer Staubbelastung (Aluminium, Quarz) können auch sekundäre Alveolarproteinosen auftreten.[3]
Befunde
Auf der Röntgenaufnahme des Thorax zeigen sich beidseitige symmetrische Infiltrationen, ähnlich einem interstitiellen Lungenödem oder einer diffusen Lungengerüsterkrankung. Auch schmetterlingsförmige Veränderungen werden beobachtet.
Die hochauflösende Computertomographie zeigt wegweisende dichte alveoläre Infiltrate mit deutlichen Bronchopneumogrammen.
Die Lungenfunktion geht mit schweren Störungen des Gasaustausches und Hypoxämie, erniedrigtem CO-Transferfaktor und Zeichen pulmonaler Shunts bei mäßiger restriktiver Ventilationsstörung einher, ohne dass wesentliche Obstruktion beobachtet werden.
Tumormarker und Surfactant-Merkmale sind in spezifischen Mustern verändert. Im Schauglas zeigt bronchoalveoläre Lavage eine milchig-trübe Flüssigkeit mit PAS-positivem Sediment, was mit seiner Mehrlagigkeit ein typisches Merkmal ist.
In fortgeschrittenen Fällen findet sich eine Polyglobulie und Erhöhung der Lactatdehydrogenase (LDH).
Therapie
Eine medikamentöse Therapie des ursächlichen Krankheitsgeschehens ist unbekannt, behandelt wird nur, wenn bedeutsame Symptome auftreten, dann wird bei einer primären Alveolarproteinose eine therapeutische bronchoalveoläre Lavage durchgeführt. Diese wird mit vielen Litern isotoner Spülflüssigkeit in Vollnarkose vorgenommen.
Genetik
Die Erkrankung hat zumindest in der kongenitalen Form oder im Kindesalter genetische Ursachen. Bei weißen Siedlern auf der Insel Réunion tritt die Erkrankung in der kindlichen Form gehäuft auf, was die Erforschung dieser genetischen Grundlagen befördert hat.
Literatur
- Matthias Griese: Molekulare Grundlagen und Klinik der pulmonalen Alveolarproteinosen. In: Dtsch Arztebl. 2002; 99, S. A 1013–1023 [Heft 15]
- Joachim Müller-Quernheim: Interstitielle Lungenerkrankungen. Thieme, Stuttgart 2003, ISBN 3-13-132281-0.
Einzelnachweise
- J. F. Seymour, J. J. Presneill: Pulmonary alveolar proteinosis: progress in the first 44 years. In: Am. J. Respir. Crit. Care Med. Band 166, Nr. 2, Juli 2002, S. 215–235, doi:10.1164/rccm.2109105, PMID 12119235.
- S. H. Rosen, B. Castleman, A. A. Liebow: Pulmonary alveolar proteinosis. In: New England Journal of Medicine. 1958; 258, S. 1123–1142. PMID 13552931.
- W. Böcker, H. Denk, Ph. U. Heitz, H. Moch: Pathologie. 4., vollst. überarb. Auflage, München 2008, ISBN 978-3-437-42382-6, S. 627.