β-Mannosidose
Die β-Mannosidose ist eine äußerst seltene autosomal-rezessiv vererbte lysosomale Speicherkrankheit aus der Gruppe der Oligosaccharidosen.
| Klassifikation nach ICD-10 | |
|---|---|
| E77.1 | Defekte beim Glykoproteinabbau Mannosidose |
| ICD-10 online (WHO-Version 2019) | |
Prävalenz
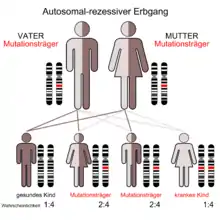
Die β-Mannosidose ist mit einer Prävalenz von kleiner als 1 : 1.000.000 bei lebendgeborenen Kindern eine ausgesprochen seltene Erkrankung. Weltweit wurden bis 2008 nur 20 Fälle in 16 Familien berichtet.[1] Aufgrund der schlechten Datenlage sind die Symptome nicht ausreichend gesichert beschrieben.[2]
Symptome
Ähnlich der α-Mannosidose ist die β-Mannosidose bei den betroffenen Patienten durch Immunschwäche, geistige Retardierung, verzögerte Sprachentwicklung, Veränderungen am Skelett und Schwerhörigkeit gekennzeichnet. Hinzu kommen in einigen Fällen Hypotonie und Krampfanfälle. Das geschwächte Immunsystem hat rezidivierende Atemwegsinfekte zur Folge.[2]
Genetik und Pathogenese
Der β-Mannosidose liegt ein autosomal-rezessiver Erbgang zugrunde. Mutationen im MANBA-Gen, das sich auf Chromosom 4 Genlocus q22-q25 befindet, sind die Ursache der Erkrankung. Das MANBA-Gen codiert für das Enzym β-Mannosidase. Mutationen in diesem Genprodukt können eine verminderte Aktivität der β-Mannosidase bewirken, wodurch sich im Gewebe der betroffenen Patienten Disaccharide aus Mannose anreichern.
Diagnose
Die Diagnose kann durch die Bestimmung der Aktivität der β-Mannosidase in Leukozyten oder kultivierten Fibroblasten gestellt werden. Eine DNA-Analyse (‚Gentest‘) kann zur Bestätigung der Ergebnisse durchgeführt werden. Die Ausscheidung erhöhter Mengen von Mannose-Disaccharid über den Urin ist ein Indiz für die Erkrankung, aber kein spezifischer Nachweis. Eine pränatale Diagnose ist sowohl biochemisch als auch molekulargenetisch möglich.[2]
Therapie
Eine spezifische Therapie ist bisher nicht bekannt. Die Krankheit wird rein symptomatisch behandelt.
Erstbeschreibung und Veterinärmedizin
Die Erkrankung wurde 1981 erstmals bei Ziegen beschrieben,[3] und fünf Jahre später erstmals beim Menschen.[4] Der Krankheitsverlauf in Ziegen ist deutlich ernsthafter und führt zu einem frühen Tod der Tiere.[3] Bei Rindern wurde die Erkrankung erstmal 1993 festgestellt.[5]
Weiterführende Literatur
- M. Zhu u. a.: Beta-mannosidosis mice: a model for the human lysosomal storage disease. In: Hum Molec Genet 15, 2006, S. 493–500, PMID 16377659.
- J. R. Leipprandt u. a.: Caprine beta-mannosidase: sequencing and characterization of the cDNA and identification of the molecular defect of caprine beta-mannosidosis. IN: Genomics 37, 1996, S. 51–56, PMID 8921369.
- T. Levade u. a.: Human beta-mannosidase deficiency associated with peripheral neuropathy. In: Ann Neurol 35, 1994, S. 116–119, PMID 8285582.
- J. A. Malachowski und M. Z. Jones: Beta-mannosidosis: lesions of the distal peripheral nervous system. In: Acta Neuropath 61, 1983, S. 95–100, PMID 6637401.
- K. L. Lovell und M. Z. Jones: Distribution of central nervous system lesions in beta-mannosidosis. In: Acta Neuropath 62, 1983, S. 121–126, PMID 6659869.
Weblinks
- Β-Mannosidose. In: Online Mendelian Inheritance in Man. (englisch)
- Β-Mannosidose. In: Orphanet (Datenbank für seltene Krankheiten).
Einzelnachweise
- H. M. F. Riise Stensland u. a.: Identification of two novel beta-mannosidosis-associated sequence variants: biochemical analysis of beta-mannosidase (MANBA) missense mutations. In: Molec Genet Metab 94, 2008, S. 476–480, PMID 18565776.
- Β-Mannosidose. In: Orphanet (Datenbank für seltene Krankheiten).
- M. Z. Jones und G. Dawson: Caprine beta-mannosidosis: inherited deficiency of beta-D-mannosidase. In: J Biol Chem 256, 1981, S. 5185–5188, PMID 7228876.
- D. A. Wenger u. a.: Human beta-mannosidase deficiency. In: NEJM 315, 1986, S. 1201–1205, PMID 2945113.
- M. Z. Jones und B. Abbitt: Animal model of human disease: bovine beta-mannosidosis. In: Am J Path 142, 1993, S. 957–960, PMID 8456950.