Rieger-Syndrom
Der Begriff Rieger-Syndrom (Syn. Irido-Dentale-Dysplasie, Axenfeld-Syndrom, Rieger-Axenfeld-Syndrom und Dysgenesis mesodermalis corneae et iridis) bezeichnet eine Hemmungsmissbildung des Mesoderms aufgrund einer Genmutation.[1][2] Das Syndrom wird autosomal-dominant vererbt. Betroffen sind die Chromosomen 4, 6, 11 und 18, konkret sind derzeit die Genorte 4q25-27, 13q14, 6p25 und 6q24 bekannt.[1][3][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q13.8 | Sonstige angeborene Fehlbildungen des vorderen Augenabschnittes Rieger-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
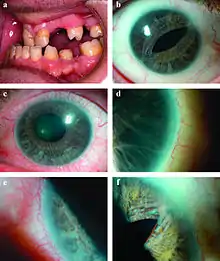
Klinisches Bild
Typisch ist eine Irisdysplasie, die als Kolobom, Lochbildung, Verformung der Pupille oder Synechien imponiert. Sie kann, meist in Verbindung mit Fehlbildungen im Kammerwinkel des Auges zum Glaukom führen.[1] Der Pathomechanismus für die Entstehung des Glaukoms beim Rieger-Syndrom ist jedoch variabel.[3]
Als schwerwiegende weitere Symptome sind Mittelohrschwerhörigkeit, cerebrale Retardierung, Zahn-, Kiefer- und Gesichtsschädelfehlbildungen (Mikrodontie, verminderte Zahnzahl siehe Hypodontie), Nabelhernie, und Knochenbildungsstörungen am Skelettsystem bekannt.[1][3][2]
Differentialdiagnostik
Abzugrenzen ist u. a. die Peters-Anomalie.
Therapie
Eine ursächliche Therapie ist bislang nicht bekannt, mit unbefriedigendem Erfolg sind symptombezogene plastisch-operative Korrekturen möglich und insbesondere im Bezug auf die Sehkraft nötig.[1][3]
Siehe auch
Einzelnachweise
- R. Witkowski u. a.: Lexikon der Syndrome und Fehlbildungen. Springer, 2003, ISBN 3-540-44305-3, S. 1107, (online)
- A. Burk: Checkliste Augenheilkunde. Thieme Verlag, 2005, ISBN 3-13-100573-4, S. 547, (online)
- D. V. Michalk: Differentialdiagnose Pädiatrie. Urban & FischerVerlag, 2005, ISBN 3-437-22530-8, S. 187, (online)