Myelolipom
Myelolipome sind seltene gutartige Tumoren oder tumorähnliche Läsionen, die sich aus reifem Fettgewebe und variablen Mengen hämatopoetischen Gewebes zusammensetzen und sich meist im Bereich der Nebenniere manifestieren.[1][2] Der Begriff Myelolipom wurde 1929 von dem französischen Pathologen Charles Oberling (1895–1960) geprägt.[3]
| Klassifikation nach ICD-O-3 | |
|---|---|
| 8870/0 | Myelolipom |
| ICD-O-3 erste Revision online | |
Epidemiologie
In Obduktionsstudien wurden Myelolipome bei 0,08–0,4 % der Fälle gefunden. Männer und Frauen sind etwa gleichhäufig betroffen. Der Altersgipfel liegt in der 5.–7. Lebensdekade.[1]
Ätiologie
Die der Entstehung von Myelolipomen zugrunde liegenden Ursachen sind unbekannt. Diskutiert wird die Möglichkeit einer Metaplasie retikuloendothelialer Zellen von Blutkapillaren infolge von Stimuli wie Nekrose, Infektion oder Stress. Mitunter wird der Tumor auch als Ort einer extramedullären Blutbildung aufgefasst.[1] Neuere Untersuchungen zeigen, dass sowohl die Fett- wie auch die myeloide Komponente monoklonalen Ursprungs sind, so dass es sich bei dem Myelolipom möglicherweise um eine echte Neoplasie handelt.[4] In der Literatur wird auch eine Assoziation mit einer angeborenen Nebennierenhyperplasie beschrieben.[5]
Pathologie
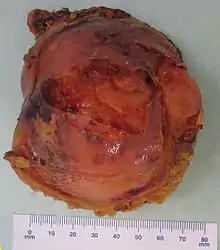
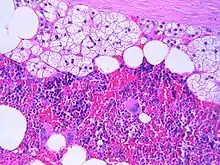
Myelolipome sind gelbe bis bräunliche, wenige Millimeter bis 30 cm große, umschriebene, jedoch nicht gekapselte Tumoren. Sie treten üblicherweise solitär und einseitig im Bereich einer Nebenniere auf, werden selten jedoch auch bilateral und/oder außerhalb der Nebenniere (z. B. im Retroperitoneum, Mediastinum, der Leber, in Muskelfaszien) gefunden. Mikroskopisch setzt sich der Tumor aus reifem Fettgewebe und myeloiden Zellen zusammen. Gelegentlich werden Infarktareale, Einblutungen oder knöcherne Metaplasien beobachtet.[1][2]
Symptomatik
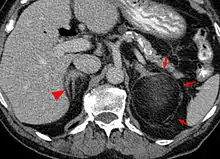
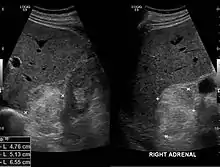
Die Mehrzahl der Myelolipome sind asymptomatisch und werden zufällig, etwa im Rahmen radiologischer Untersuchungen oder einer Obduktion gefunden. Nur gelegentlich, insbesondere bei größeren Tumoren, berichten betroffene Patienten von Bauch- oder Flankenschmerz. Selten bestehen endokrine Störungen wie das Cushing-, Conn-Syndrom oder eine angeborene Nebennierenhyperplasie.[1]
Therapie
Kleine, asymptomatische Myelolipome bedürfen keiner Therapie. Symptomatische Tumoren werden durch eine chirurgische Entfernung der Nebenniere behandelt.[1]
Prognose
Die Prognose von Myelolipomen ist üblicherweise gut. Eine maligne Entartung des Tumors wurde bislang nicht berichtet. Als seltene Komplikation kann eine spontane oder traumatisch bedingte Tumorruptur mit folgender Blutung eintreten. Daten zur Mortalität bei Myelolipomen sind aufgrund der Seltenheit der Läsion nicht verfügbar.[1]
Quellen
- Wegener: Ganzkörpercomputertomographie. Blackwell Wissenschaft, Berlin 1992, ISBN 3-89412-105-X.
- Michael Federle: Abdomen: Die 100 Top-Diagnosen. Urban & Fischer Verlag, 2004, ISBN 3-437-23590-7, S. 319–321.
- M. Goepel u. a.: Lipom der Nebenniere, Fallbericht und Literaturübersicht. In: Urologe. 37, 1998, S. 526–539.
Einzelnachweise
- P. Ramchandani: Adrenal Myelolipoma. (15. Februar 2007). (online auf Medscape)
- C. D. M. Fletcher: Diagnostic Histopathology of Tumors. 3. Auflage. Vol. 2., Churchill Livingstone, 2007.
- Oberling: Les formations myelolipomaleuses. In: Bull. Cancer. 1929;18, S. 234.
- A. McNicol: A diagnostic approach to adrenal cortical lesions. In: Endocr Pathol. (2008) 19, S. 241–251. PMID 19089656
- A. Reineke-Lüthge, F. Koschoreck, G. Klöppel: The molecular basis of persistent hyperinsulinemic hypoglycemia of infancy and its pathologic substrates. In: Virchows Arch. 2000 Jan;436(1), S. 1–5. PMID 10664155