Gallengangszyste
Gallengangszysten sind angeborene Fehlbildungen der Gallengänge, die zur Erweiterung der Gallengänge führen. Sie sind in westlichen Ländern selten,[1] treten jedoch häufiger in Asien wie Japan und China auf.
| Klassifikation nach ICD-10 | |
|---|---|
| Q44.4 | Gallenganszyste |
| ICD-10 online (WHO-Version 2019) | |
Symptome
Die Krankheit zeigt sich häufig im ersten Lebensjahr. Nur selten tritt sie erst im Erwachsenenalter auf und ist dann gewöhnlich mit einer erhöhten Komplikationsrate verbunden. Die klassische Trias aus Bauchschmerzen, Gelbsucht und einer Verhärtung im rechten oberen Quandranten des Bauches findet sich nur bei wenigen Patienten.
Diagnose
Die Diagnose kann anhand von Anamnese, Palpation und Sonografie gestellt werden. Bei der Blutuntersuchung ist direktes oder konjugiertes Bilirubin erhöht.
Typen
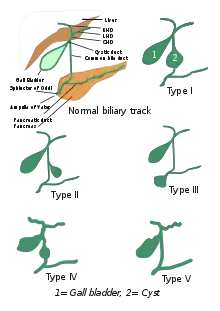
Die Gallengangszysten werden nach Todani 1977 in fünf verschiedene Klassen unterteilt.[2]
Die Klassifikation basiert auf der Stelle des Auftretens der Zyste. Unterteilt wird von Typ I bis Typ IV.
- Typ I: Am häufigsten auftretend (80–90 %), gekennzeichnet durch sakkuläre oder fusiforme Aussackungen eines Teils oder des gesamten Ductus choledochus.
- Typ II: Isolierte extrahepatische Aussackung des Gallenganges.
- Typ III oder Choledochozele: Ausgehend vom duodenalen Abschnitt des Gallenganges oder der Schnittstelle mit dem Ductus pancreaticus.
- Typ IVa: Charakterisiert durch mehrere Aussackungen und/oder des intrahepatischen und extrahepatischen Gallenganges.
- Typ IVb: Mehrere Aussackungen die nur den extrahepatischen Gallengang betreffen.
- Typ V: Zystische Aussackung des intrahepatischen Gallenganges.
Therapie
Gallengangszysten werden chirurgisch behandelt. Die Zyste wird entfernt und es wird eine Roux-en-Y-Anastomose des Gallenganges geschaffen. Die Komplikationen beinhalten eine Entzündung der Gallengänge (Cholangitis) und einer malignen Entartung (2 %), welche an jeglicher Stelle der Gallengänge sich bilden kann. Ein kürzlich erschienener Artikel beschreibt auch die Methode die Zyste laparoskopisch zu entfernen und anschließend die Gallengänge über eine Roux-Y-Anastomose von der Leber zum Jejunum führen zu lassen.[3] Im Falle einer Gallengangszyste vom sakkulären Typ wird die Zyste chirurgisch entfernt ein T-förmiger Tubus platziert.
Weblinks
- Resection of a Type I Choledochal Cyst Video. The Toronto Video Atlas of Liver, Pancreas and Transplant Surgery
Einzelnachweise
- Y. B. Liu, J. W. Wang, K. R. Devkota u. a.: Congenital choledochal cysts in adults: twenty-five-year experience. In: Chin. Med. J. Band 120, Nr. 16, 2007, S. 1404–1407, PMID 17825168.
- T. Todani, Y. Watanabe, M. Narusue, K. Tabuchi, K. Okajima: Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. In: The American Journal of Surgery. Band 134, Nr. 2, 1977, S. 263–269, doi:10.1016/0002-9610(77)90359-2, PMID 889044.
- M. Diao, L. Li, Q. Li, M. Ye, W. Cheng: Single-incision versus conventional laparoscopic cyst excision and Roux-Y hepaticojejunostomy for children with choledochal cysts: a case-control study. In: World Journal of Surgery. Band 37, Nummer 7, Juli 2013, ISSN 1432-2323, S. 1707–1713, doi:10.1007/s00268-013-2012-y, PMID 23539195.