Insulin im Gehirn
Insulin ist als Hormon des Energiestoffwechsels besonders für die Regulation des peripheren Glukosemetabolismus und Förderung anaboler Vorgänge zuständig. Es ist zwar schon länger bekannt, dass Insulin und auch der Insulinrezeptor ebenfalls im Zentralnervensystem auftreten, jedoch ist ihre dortige Funktion noch immer nicht vollständig geklärt. Komplexe Auswirkungen auf Neurone und Kognition werden immer deutlicher und gewinnen dabei an Bedeutung.
Insulinquellen
Der Insulingehalt im Gehirn ist zwischen 10 und 100 Mal höher als im Blutplasma. Die Menge des Insulins im Gehirn schwankt zudem während der Entwicklung: Das fetale Gehirn besitzt einen höheren Insulinspiegel als das Gehirn eines Erwachsenen.
Periphere Insulinquellen
Das meiste Insulin im Körper wird durch die β-Zellen des Pankreas gebildet. Dieses im Blutkreislauf zirkulierende Insulin kann die Blut-Hirn-Schranke passieren und das Zentralnervensystem erreichen. Ein akut ansteigender peripherer Insulinspiegel wirkt sich jedoch kaum auf den Insulingehalt im Gehirn aus.
Neuronale Insulinquellen
Eine geringe Menge an Insulin wird auch im Gehirn durch Pyramidal-Neurone synthetisiert. Einige GABAerge Neurone weisen sogar eine transmitterartige Freisetzung des Insulins auf. Gliazellen scheinen dagegen kein Insulin zu produzieren. Die tatsächliche Rolle des neuronal synthetisierten Insulins und sein Anteil an der Gesamtinsulinmenge im Gehirn ist jedoch weiterhin umstritten.
Insulin und die Blut-Hirn-Schranke
Die Blut-Hirn-Schranke verfügt über spezielle Insulin-Bindungsstellen. Dabei lassen sich zwei verschiedene Funktionen ausmachen: zum einen die klassische Rezeptorfunktion (Signalübertragung) und zum anderen die Transportfunktion. Es ist jedoch noch nicht geklärt, ob es sich um Proteine gleichen Gen-Ursprungs oder um jeweils einzigartige Proteine handelt.
Rezeptorfunktion
Die Bindungsstellen, die der Signalübertragung dienen, wirken sich auf die Zellen der Blut-Hirn-Schranke aus (BEC = Brain Endothelial Cells). So werden durch Insulinwirkung die Transportprozesse von Leptin, Tyrosin, Azidothymidin (ein Mittel zur AIDS-Behandlung) verbessert und die Genexpression von P-Glykoprotein sowie der katalytischen Untereinheit der Glutamatcysteinligase gesteigert. Am Plexus choroideus kann die Insulinwirkung möglicherweise die Produktion des Liquor cerebrospinalis beeinflussen. Zudem enthalten die BEC, vor allem auf der Gehirn zugewandten Membran, das Insulysin (IDE = Insulin-degrading enzyme) und können somit evtl. die Insulinwirkung sowie Insulintransport regulatorisch beeinflussen.
Transportprozess
Der Insulintransport durch die Blut-Hirn-Schranke erfolgt über einen rezeptorvermittelten, aktiven Transportmechanismus. Da ein akuter Anstieg des im Blut vorhandenen Insulins sich kaum auf den Insulingehalt im Gehirn auswirkt, handelt es sich bei dem Transport um einen sättigbaren Transportvorgang. Bei Neugeborenen ist die Insulin-Bindung noch deutlich höher als in Erwachsenen.
Bemerkenswert ist die Tatsache, dass der Transport von Insulin durch die Blut-Hirn-Schranke je nach Hirnregion variiert: die höchste Permeabilität findet man im Bereich der Pons, Medulla oblongata und des Hypothalamus, wohingegen der occipitale Cortex die niedrigste Permeabilität aufweist. Im Bereich des Thalamus und Mesencephalon ist die Blut-Hirn-Schranke sogar vollständig impermeabel für Insulin.
Der Transportprozess wird zudem durch eine ganze Reihe von Faktoren beeinflusst: Hungern, Adipositas, Eisenstatus, Stickstoffmonoxid-Level und Glukokortikoide. Während die Glukokortikoide den Transport vermindern, wirkt sich Stickstoffmonoxid (NO) auf unterschiedliche Weisen aus: NO, welches durch nNOS produziert wurde, inhibiert den Insulintransport; NO, welches durch eNOS oder iNOS produziert wurde, stimuliert jedoch den Transportprozess. Da sich das Stickstoffmonoxid selbst nicht voneinander unterscheidet, wird einer der beiden Wege seine Wirkung indirekt über andere Zellen der neurovaskulären Einheit ausüben.
Neuronaler Insulinrezeptor und Signalwege
Verteilung im Gehirn
Die Verteilungsdichte des neuronalen Insulinrezeptors (nIR) im Gehirn kann zwischen den verschiedenen Hirnregionen um das 5- bis 10-Fache variieren. Dabei ist die Verteilung unabhängig von Vaskularisierung und Zelldichte, jedoch besonders hoch in dendritischen Feldern mit starkem synaptischen Input. Letzteres weist auf einen möglichen Zusammenhang zwischen neuronaler Aktivität und der Verteilungsdichte des nIR hin. Regionen mit hoher nIR Verteilungsdichte sind: Neocortex, Hippocampus, Bulbus olfactorius, Hypothalamus und Cerebellum.
Während der Entwicklung vom embryonalen bis zum adulten Gehirn unterliegt die nIR-Dichte einer stetigen Veränderung. Einige Hirnregionen wie z. B. Striatum und Thalamus weisen während der Entwicklungsphasen eine hohe nIR Dichte auf, die sich später im adulten Gehirn verringert.
Verteilung auf zellulärer Ebene
Der nIR ist vor allem in den pre- und postsynaptischen Membranen der Neurone lokalisiert. Passend dazu sind auch die IRS (z. B. IRSp58 oder IRSp53; siehe unten: Signalwege) besonders in den Synapsen von Neocortex, Hippocampus und Cerebellum konzentriert. Die Synapsen sind somit ein wichtiger Angriffspunkt der Insulinsignalisierung im Gehirn.
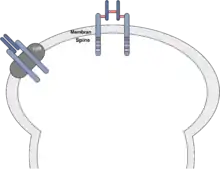
Zusätzlich reagieren die nIR in der Zellmembran nur dann auf Insulin, wenn sie außerhalb von Ganglioside GM1 – rafts liegen. Innerhalb dieser Membranabschnitte werden die nIR durch eine hohe Tyrosin-Phosphatase Aktivität gehemmt. Ein verändertes Verhältnis von Cholesterol und Sphingolipiden in den Dornfortsätzen (dendritic spines) ermöglicht außerdem die Modifizierung von Rezeptoraktivitäten inklusive des nIR.
Struktur und Unterscheidung der Insulinrezeptoren
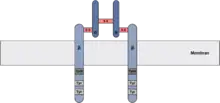
Der Insulinrezeptor (IR) gehört zur Familie der Rezeptortyrosinkinasen. Er ist ein tetrameres Glykoprotein, bestehend aus zwei α-Untereinheiten und zwei β-Untereinheiten die durch Disulfidbindungen zusammengehalten werden. Die α-Untereinheiten liegen vollständig extrazellulär und binden das Insulin sowie mit geringerer Affinität auch IGF-1. Die β-Untereinheiten besitzen eine extrazelluläre, eine transmembran und eine intrazelluläre Domäne.
Grundsätzlich besitzen der neuronale IR (auch IR-A) und der periphere IR (auch IR-B) identische pharmakologische und kinetische Eigenschaften. Der nIR weist jedoch eine geringere Glykosylierung auf und unterscheidet sich somit in der Kohlenhydrat-Zusammensetzung und der molekularen Größe vom peripheren IR. Durch alternatives Spleißen fehlt dem nIR zudem das Exon 11.
Unterschiede finden sich auch in der Regulation durch Insulin: der periphere IR wird durch Insulin downreguliert (die Bindung eines weiteren Insulins am Rezeptordimer löst das Erste), der nIR unterliegt dieser Downregulierung jedoch nicht.
Am bedeutendsten ist jedoch die Unterscheidung der beiden IR aufgrund ihrer Funktion: während der periphere IR für seine schnellen metabolischen Effekte, v. a. durch Insertion von Glukosetransportern, bekannt ist, wirkt sich der nIR sehr komplex auf die Neurone aus und bewirkt kurz- wie auch langfristige Veränderungen (siehe unten: Physiologische Funktionen).
Bemerkenswert ist jedoch, dass im Zentralnervensystem beide Insulinrezeptoren gefunden werden. Neben den Neuron-spezifischen nIR tritt der periphere IR in geringer Dichte in einigen Gliazellen auf.
Signalwege
Der neuronale Insulinrezeptor wirkt prinzipiell, genau wie der periphere IR, vor allem über die beiden PI3K/Akt- und Ras/MAPK-Signalwege. Das tatsächliche downstream Signal kann sich jedoch selbst zwischen einzelnen Neuronen je nach Verfügbarkeit der downstream Substrate unterscheiden. Weitere Signalwege und auch Cross-talking zwischen verschiedenen Signalkaskaden sind bisher nur wenig erforscht, werden aber in Zukunft sicher an Bedeutung gewinnen.
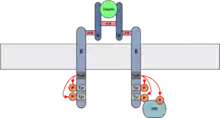
Beginn der Signaltransduktion
Die Bindung von Insulin an der α-Untereinheit führt zur Aktivierung der Tyrosinkinaseaktivität der β-Untereinheit, welche zunächst die eigenen Tyrosin-Reste autophosphoryliert. Anschließend phosphoryliert die β-Untereinheit auch die Tyrosin-Reste eines Insulin-Rezeptor-Substrates (IRS). Von dem IRS gehen dann die verschiedenen Signalwege aus. Das IRS-1 und IRS-2 vermitteln dabei die meisten pleiotropen Effekte in den verschiedenen Zellen.
PI3K/Akt-Weg
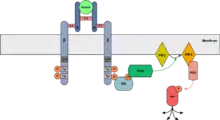
Der PI3K/Akt-Signalweg beginnt mit der Bindung der p85-SH2-Domäne des PI3K am phosphorylierten Tyrosin des aktivierten IRS. Durch diese Bindung aktiviert, phosphoryliert die p110-Untereinheit des PI3K das membranständige PIP2 zu PIP3. Das somit veränderte Membranphospholipid bindet die PDK (phosphoinositide-dependent kinase), das wiederum Akt (PKB) durch Phosphorylierung aktiviert. Von dem Akt gehen anschließend weitere vielfältige Effekte aus.
Ras/MAPK-Weg
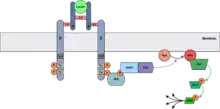
Der Ras/MAPK-Signalweg beginnt mit der Bindung von Grb2 mit seiner SH2-Domäne zum phosphorylierten Tyrosin des aktivierten IRS. An das gebundene Grb2 bindet sich wiederum das SOS das anschließend Ras durch den Austausch von GDP zu GTP aktiviert. Das aktive Ras bindet und aktiviert Raf-1 und beginnt die Kinase-Kaskade mit der Phosphorylierung von MEK und der darauf folgenden Phosphorylierung von ERK. Das aktivierte ERK gelangt in den Zellkern und beeinflusst dort über Transkriptionsfaktoren die Genexpression.
PKC-/NF-kB-Weg
Die Expression des P-Glykoproteins in den BEC (siehe oben: Insulin und Blut-Hirn-Schranke) wird über den für die Insulinrezeptoren untypischen PKC/NF-κB-Signalweg beeinflusst. Viele Überlappungen in den Mechanismen der PKC- und PI3K-Wege wie z. B. die Aktivierung von PKC durch PDK weisen auf weiteres Cross-talking hin. Es ist also falsch, die Signalwege als eigenständig und isoliert voneinander zu betrachten.
Physiologische Funktionen
Zentraler Glukosemetabolismus
Der Glukosemetabolismus im Zentralnervensystem wird üblicherweise, mit den Glukosetransportern GLUT-3 für Neurone und GLUT-1 für Astrozyten, als insulinunabhängig betrachtet. Dennoch weisen einige Neurone z. B. im Hippocampus den insulinabhängigen GLUT-4 überlappend mit dem Insulinrezeptor auf. Auch im Cerebellum beeinflusst Insulin in einigen Zellen die Expression des GLUT-4.
Ein weiterer Mechanismus über den Insulin den Glukosehaushalt im Gehirn beeinflussen kann, ist die Inhibition der neuronalen Adrenalin-Aufnahme. Die darauffolgende Aktivierung glialer β-Adrenorezeptoren führt zur Glukose-Sekretion aus dem Glykogenvorrat der Astrozyten.
Peripherer Glukosemetabolismus
Die Insulinwirkung im Hypothalamus kann sich auch auf die Glukoseproduktion in der Leber auswirken und diese unterdrücken. Eine defiziente Insulinsignalisierung im Hypothalamus führt zudem zu einer Desensibilisierung der Leber gegenüber zirkulierendem Insulin und somit zu einer gesteigerten Gluconeogenese.
Wirkung auf Feuerrate und Membranpotenzial
Die Wirkung von Insulin auf die Feuerrate und das Membranpotenzial der Neurone ist sehr komplex und kann sich von Zelle zu Zelle stark unterscheiden. Allgemein zeigt Insulin jedoch einen stimulatorischen Effekt auf die Na+/K+-ATPase des Gehirns.
Im Cortex der Insula wirkt sich Insulin über den PI3K-Weg negativ auf das Schwellenpotential aus und erhöht somit die Feuerrate. Im Bulbus olfactorius kann die Insulinwirkung den Auswärtsstrom von spannungsabhängigen K.v 1.3 Kanälen akut durch Phosphorylierung unterdrücken. Hypothalamische Neurone werden über die Aktivierung von ATP-abhängigen K+-Kanälen hyperpolarisiert. Auf GABA-induzierte Ströme, unter anderem im Cerebellum, wirkt sich das Insulin je nach Menge und Expositionszeit inhibitorisch oder fördernd aus.
Eine Auswirkung auf die Ausschüttung von Transmitter-Vesikeln erfolgt durch intrazelluläre Erhöhung des Ca2+-Spiegels durch Insulin. Es ist jedoch offen, ob sich der insulinsensitive Ca2+-Speicher von anderen Ca2+-Lagern im Neuron unterscheidet.
Wirkung auf Rezeptoren
Die Beeinflussung der glutamatergen Transmission erfolgt über die Phosphorylierung der NR2A und NR2B Untereinheiten der NMDA-Rezeptoren. Die Folge ist eine Potenzierung der Rezeptorantworten. Durch Beeinflussung der Exozytose kann Insulin zudem die Translokation von NMDA-Rezeptoren in die Zellmembran fördern. Das Entwicklungsstadium des Gehirns wirkt sich allerdings auch auf die Insulinsignalisierung aus: während in einem embryonalen Gehirn die Entwicklung von stillen Synapsen zu funktionalen Synapsen durch hochregulation der AMPA-Rezeptoren gefördert wird, fördert die Insulinwirkung im adulten Gehirn die Clathrin-abhängige Endozytose der AMPA-Rezeptoren und somit das LTD (long-term depression).
Die Beeinflussung der GABAergen Transmission geschieht ebenfalls durch die Translokation von GABAA-Rezeptoren zur Plasmamembran. Zudem steigert Insulin auch die Expression der GABAA-Rezeptoren in postsynaptischen Membranen und somit die Amplituden der mIPSC’s (miniature inhibitory postsynaptic current). Die Verteilungsdichte der GABAA-Rezeptoren auf der postsynaptischen Membran wird zusätzlich noch durch Akt abhängige Phosphorylierung reguliert.
Weitere neuromodulatorische Effekte
Die Insulinsignalisierung wirkt sich ebenfalls relativ diffus auf das monoaminerge System aus. So kann Insulin über den PI3K-Signalweg die Expression der Dopamintransporter erhöhen und die Aktivität der Monoaminoxidase, welche Serotonin und Dopamin degradiert, verringern. Auch die Fluktuationsrate von Adrenalin und Noradrenalin kann durch Hemmung der Wiederaufnahme erhöht werden. Im Hypothalamus werden durch Insulin selektiv die α2-adrenergen Rezeptoren runterreguliert.
Anderweitige Effekte sind unter anderem die Stimulation der Aminosäuren-Aufnahme durch Neurone zur Neurotransmitter Synthese oder die Erhöhung des Phosphatidylinositol-Gehaltes in der Membran hippocampaler Neurone. Letzteres ist ein weiterer Weg, durch den Insulin die rezeptorvermittelte Signaltransduktion beeinflussen kann.
Proliferation
Die Proliferation und Differenzierung von multipotenten neuronalen Stammzellen stehen offensichtlich auch unter dem Einfluss der von Insulin induzierten Signaltransduktion. So führt der Insulinentzug zu einem nicht-apoptischen, autophagischen Zelltod und auch das Fehlen des IRS-2 beeinträchtigt die neuronale Proliferation. Umgekehrt erhöht Insulin die Aktivität der Ornithin-Decarboxylase, welche ein Indikator für Wachstums- und Differenzierungsvorgänge ist.
Differenzierung
Während der Zelldifferenzierung in dem entwickelnden Gehirn steigen die Level von Insulin und Insulinrezeptoren. Die damit einhergehende veränderte Bindungsrate des Insulins an die Plasmamembran stimmt mit den Änderungen des Gehirnwachstumes überein. Sie erreicht ihren Höhepunkt kurz nach der Geburt und verringert sich anschließend wieder bis zu den adulten Verhältnissen. Gerade bei diesen entwicklungsbedingten Schwankungen ist das endogen synthetisierte Insulin möglicherweise von Bedeutung. Die Insulinsignalisierung kann auch als integrierender Faktor, der die neuronale Differenzierungsgeschwindigkeit mit den Änderungen im Nahrungsangebot abstimmt, wirken und somit eine wichtige koordinative Rolle übernehmen.
Axonwachstum
Auch bei dem Axonwachstum spielt das endogene Insulin eine wichtige Rolle und übertrifft das periphere Insulin in seiner Wirksamkeit. Dabei wirkt Insulin gleich an mehreren wichtigen Stellen: die korrekte Verteilung der Neurofilamente in den Axonen und auch die gesamte Neuronenmorphologie ist auf die funktionierende MAPK-Kaskade angewiesen. Über PI3K/mTOR wird das Auswachsen von Axonen (z. B. in cerebellären Körnerzellen) gefördert und die tau-Synthese gesteigert. Das Tau-Protein wirkt sich auf Wachstum und Polarität des Axones aus. Die Tubulin-Synthese wird durch vermehrte Expression der mRNA und durch Stabilisierung dieser gefördert. Insulin und IGF-2 sind zudem notwendig, damit NGF an seinen Rezeptor binden kann. Der Insulinrezeptor wird darüber hinaus auch noch bei der Axonalen-Wegfindung benötigt.
Neuroprotektorische Effekte
Die durch Insulin angestoßenen Signalwege wirken sich auch neuroprotektorisch aus. Dabei ist es jedoch wichtig, dass einige dieser Effekte teilweise auch von Astrozyten modifiziert werden.
Gegen Apoptose
Die antiapoptischen Wirkungen des Insulins werden über viele verschiedene Signalwege ausgeübt. Über PI3K/Akt verhindert Insulin die Bcl-2 Reduzierung, Caspase-3 Aktivierung, DNA-Fragmentierung und bewirkt die Inhibition von GSK-3β. Durch Wirkung von mTOR auf das ribosomale Protein P70SK wird zusätzlich die Proteinsynthese erhalten. Bei Ascorbat/Fe2+-induziertem Zelltod erweist sich die MAPK-Kaskade als protektorischer Weg.
Ein Insulinentzug führt dagegen zu einer erhöhten Apoptoserate im Hippocampus und Hypothalamus. Während der Entwicklung sind dabei vor allem die Körnerzellen des Cerebellums betroffen. Es kommt zudem zur Umverteilung von regulatorischen Proteinen zwischen den intrazellulären Kompartimenten wie z. B. membrangebundenem Grf1 oder Reduzierung des mitochondrialen Ras.
Gegen oxidativen Stress
Die bei oxidativem Stress durch verringerte Wiederaufnahme entstehenden Akkumulationen von GABA und Glutamat können durch Insulinwirkung wieder reduziert werden. Die Glukoseaufnahme der angegriffenen GLUT-3 und die Weiterverarbeitung zu Pyruvat kann durch Insulin ebenfalls wieder stimuliert werden, sodass auch dem ATP-mangel und dem erhöhten extrazellulären ADP entgegengewirkt wird.
Zur Wiederherstellung der redox-Balance trägt der PI3K/Akt/mTOR-Weg durch Translokation des Nrf2 in den Zellkern bei. Die dadurch erhöhte Aktivität der Glutamatcysteinligase stabilisiert das GSH/GSSG Level. Auch die parallele Erhöhung der Harnsäure bringt durch Stabilisierung der Ascorbinsäure einen antioxidantischen Nutzen.
Gegen Ischämie
Insulin schützt Neurone vor Hypoxie-induzierter Nekrose und auch vor Schäden durch eine verminderte Glukosezufuhr. Jedoch ist diese Schutzfunktion nicht in allen Hirnregionen gleich stark ausgeprägt: Im Gyrus dentatus des Hippocampus entfaltet Insulin eine bessere Schutzfunktion als in den Regionen CA1 und CA3. Die durch Hypoglykämie gestörte Ca2+-Homöostase kann von hohen Dosen Insulin wieder stabilisiert werden. Die insulinbedingte Erhöhung des extrazellulären GABA-Spiegels führt zur Inhibierung der Neurone. Der dadurch verringerte Glukoseverbrauch schützt vor ischämischen Schäden, während gleichzeitig eine Laktatazidose verhindert wird. Die stimulierende Wirkung des Insulins auf die Na+/K+-ATPase erweist sich auf die gleiche Art und Weise als protektorisch. Die Verringerung des extrazellulären K+ vermindert die neuronale Feuerrate und somit auch den Energiebedarf der Zelle. Zusätzlich wird durch das gesenkte intrazelluläre Na+-Level die Akkumulation von Wasser im Neuron verhindert und wirkt somit gegen ein postischämisches Ödem.
Gedächtnis und Lernen: Effekte auf synaptische Übertragung
Lernprozesse beeinflussen die IR-Expression im Hippocampus. Während die IR-Expression in der Region CA1 steigt, sinkt sie in CA3. Das Gesamtlevel an Insulinrezeptoren nimmt jedoch ab. Da als Folge des Lernprozesses die Level von IRS-1 und Akt, sowie die Aktivität des ERK1/2 steigen, ist die Verringerung der Insulinrezeptoren durch den initialen Anstieg an IR-Aktivität bedingt.
Lernen und Kognition werden aber vor allem auch durch die neuromodulatorischen Effekte des Insulins beeinflusst. Von besonderer Bedeutung ist hier die Wirkung auf Glutamaterge und GABAerge Transmission sowie die Mitbeteiligung an Vorgängen synaptischer Plastizität wie LTP und LTD. Auch die metabolischen Auswirkungen auf die teilweise GLUT-4 expressierenden hippocampalen Neurone können hier hinzugezählt werden.
Regulation der Nahrungsaufnahme und des Körpergewichts
Die Beeinflussung der Nahrungsaufnahme erfolgt im Hypothalamus durch Insulinwirkung auf Neurone des Ncl. arcuatus. Hier hemmt Insulin die Expression der orexigenen Neuropeptide NPY (Neuropeptid Y) und AgRP (Agouti-related protein) und stimuliert die Expression der anorexigenen Neuropeptide POMC (Proopiomelanocortin) und CaRT (Cocaine and amphetamine-regulated transcript). Zusammen erhöht dies die Aktivität von α-MSH in Neuronen des Ncl. paraventricularis, wodurch die Nahrungsaufnahme gehemmt wird. Bei den orexigenen Neuronen erfolgt die Signalübertragung zumindest teilweise über den PI3K-Weg, welcher durch Beeinflussung ATP-abhängiger Kaliumkanäle zur Hyperpolarisierung des Neurons führt.
Im Gegensatz dazu kommt es bei gestörter Insulinsignalisierung zur Aktivierung von JNK, welches wiederum IRS-1 phosphoryliert und somit die feedback-Inhibition des Insulinrezeptors fördert. Die unveränderte Aktivität der orexigenen Neurone führt schlussendlich zur Gewichtszunahme.
Angiogenese im Zentralnervensystem
Die Angiogenese im ZNS wird durch sehr viele Faktoren kontrolliert und bezieht viele Zelltypen des Zentralnervensystems mit ein. Ein Schlüsselfaktor ist das durch Hypoxie aktivierte und als Transkriptionsfaktor wirkende HIF (Hypoxie-induzierter Faktor). Insulin nimmt über beide, PI3K- und MAPK-Signalwege aktivierenden Einfluss auf das HIF. Zahlreiche Gene wie z. B. EPO, VEGF, GLUT1-3 werden somit durch Hypoxie und auch durch Insulin reguliert.
Pathologische Rollen
Insulin und Morbus Alzheimer
Eine gestörte Insulinsignalisierung und eine verminderte Reaktionsbereitschaft der Insulinrezeptoren gegenüber Insulin ist ein häufiges Merkmal von an Alzheimer erkrankten Gehirnen. Eine solche Insulinresistenz kann global durch einen Diabetes Mellitus Typ-2 oder auch auf das Zentralnervensystem beschränkt auftreten. Es gibt jedoch noch weitere Faktoren, die zur gestörten Signaltransduktion beitragen: die löslichen Amyloid-β-Oligomere (AβO), welche als Synaptotoxine zum Gedächtnisverlust der Alzheimer-Erkrankung beitragen, stoßen im Gehirn ähnliche Mechanismen wie der periphere Diabetes Mellitus Typ-2 an. Über die Aktivierung des TNF-α/JNK-Signalweges wird IRS-1 sowie Akt phosphoryliert und inaktiviert. Weiterhin wird die Internalisierung und Umverteilung der Insulinrezeptoren angestoßen.
Durch die defekte Insulinsignalisierung wird wiederum die β-Amyloid Synthese begünstigt und antiapoptische Signalwege werden gehemmt. Auch die dysregulierte NMDA-Rezeptor Signalisierung und die Ausbildung von tau-Aggregaten stehen in Wechselwirkung mit den gestörten Insulin-Signalwegen.
Morbus Parkinson
Morbus Parkinson ist vor allem durch die Degeneration der dopaminergen Neurone der Substantia nigra, Pars compacta im Mesencephalon gekennzeichnet. Im gesunden Organismus weist diese Hirnregion auch eine hohe Dichte an Insulinrezeptoren auf und der IGF-1 scheint ein wirksamer protektiver Faktor gegen den Verlust von dopaminergen Neuronen zu sein.
Affektive Störungen
Mehrere Studien assoziieren Diabetes Mellitus mit depressiven Erkrankungen. Genaue Mechanismen sind zurzeit noch völlig unklar, jedoch ergeben sich mögliche Verknüpfungen bei der insulinbedingten Beeinflussung der monoaminergen Systeme oder indirekte Wechselwirkungen wie Inflammation und erhöhte Cytokinproduktion.
Quellenangaben
- W. A. Banks, J. B. Owen, M. A. Erickson: Insulin in the brain: There and back again. In: Pharmacology & Therapeutics. Band 136, 2012, S. 82–93.
- E. Blázquez, E. Velázquez, V. Hurtado-Carneiro, J. M. Ruiz-Albusac: Insulin in the Brain: Its Pathophysiological Implications for States Related with Central Insulin Resistance, Type 2 Diabetes and Alzheimer’s Disease. In: Frontiers in Endocrinology. 5, 2014, S. 161.
- F. G. De Felice, M. V. Lourenco, S. T. Ferreira: How does brain insulin resistance develop in Alzheimer’s disease? In: Alzheimer’s & Dementia. Band 10, Suppl. 1, 2014, S. S26–S32.
- A. I. Duarte, P. I. Moreira, C. R. Oliveira: Insulin in Central Nervous System: More than Just a Peripheral Hormone. In: Journal of Aging Research. 2012, S. 1–21.
- R. Ghasemi, A. Haeri, L. Dargahi, Z. Mohamed, A. Ahmadiani: Insulin in the Brain: Sources, Localization and Functions. In: Molecular Neurobiology. Band 47, 2013, S. 145–171.
- M. Gralle: The neuronal insulin receptor in its environment. In: Journal of Neurochemistry. Band 140, 2017, S. 359–367.
- S. M. Gray, R. I. Meijer, E. J. Barrett: Insulin Regulates Brain Function, but How Does It Get There? In: Diabetes. Band 63, 2014, S. 3992–3997.
- A. Kleinridders, H. A. Ferris, W. Cai, C. R. Kahn: Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. In: Diabetes. Band 63, 2014, S. 2232–2243.
- S. Kullmann, M. Heni, A. Fritsche, H. Preissl: Insulin Action in the Human Brain: Evidence from Neuroimaging Studies. In: Journal of Neuroendocrinology. Band 27, 2015, S. 419–423.
- L. Plum, M. Schubert, J. C. Brüning: The role of insulin receptor signaling in the brain. In: Trends in Endocrinology & Metabolism. Band 16, 2005, S. 59–65.
- M. Ramalingam, S.-J. Kim: Mechanisms of action of brain insulin against neurodegenerative diseases. In: Journal of Neural Transmission. Band 121, 2014, S. 611–626.
- Y. Zeng, L. Zhang, Z. Hu: Cerebral insulin, insulin signaling pathway, and brain angiogenesis. In: Neurological Sciences. Band 37, 2016, S. 9–16.